

Global landscape of SARS-CoV-2 genomic surveillance and data sharing
- PMID: 35347305
- PMCID: PMC9005350
- DOI: 10.1038/s41588-022-01033-y
Global landscape of SARS-CoV-2 genomic surveillance and data sharing
Abstract
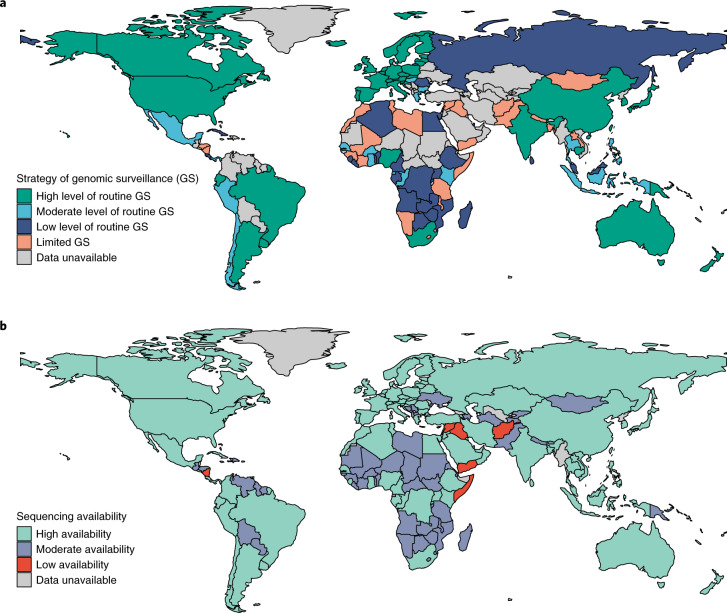
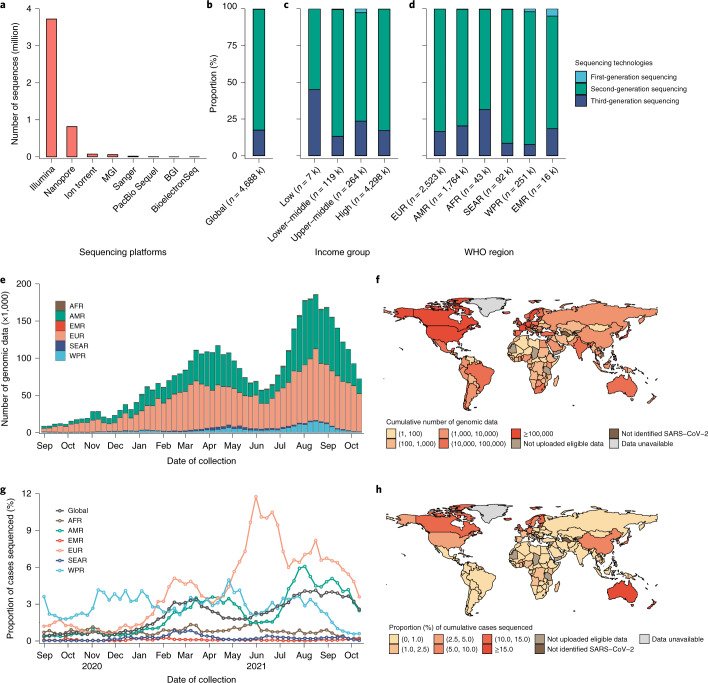
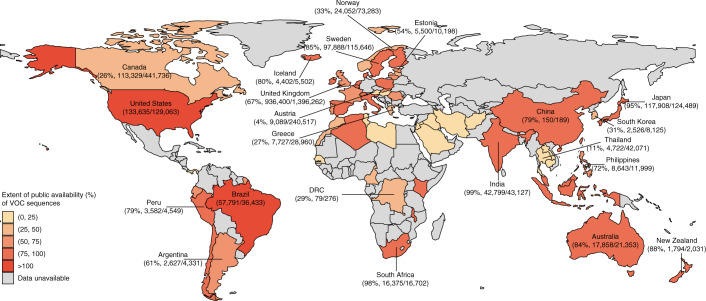
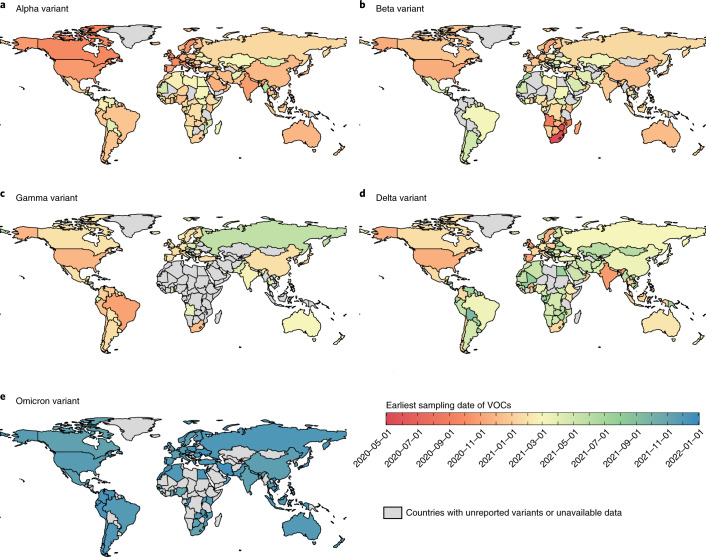
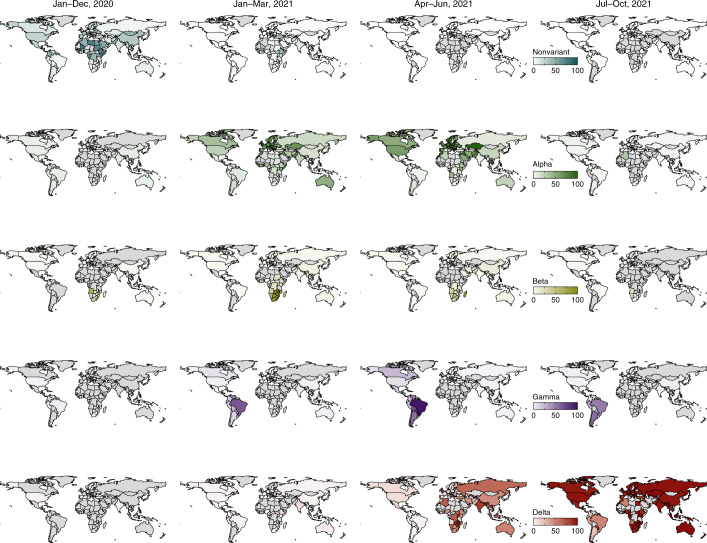
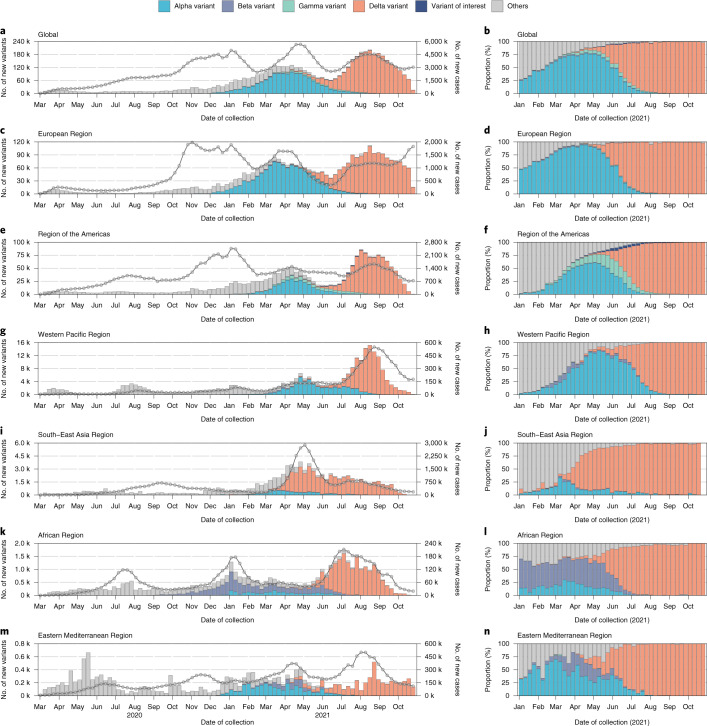
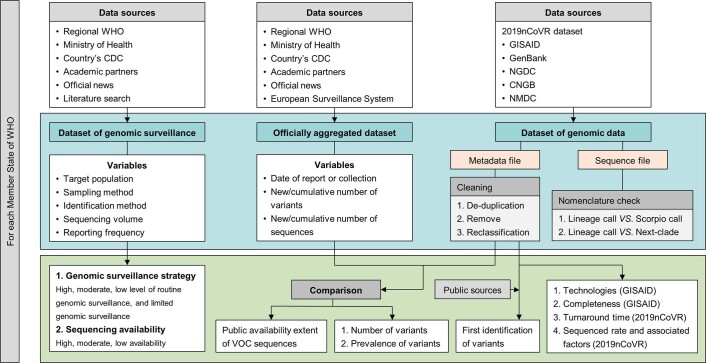
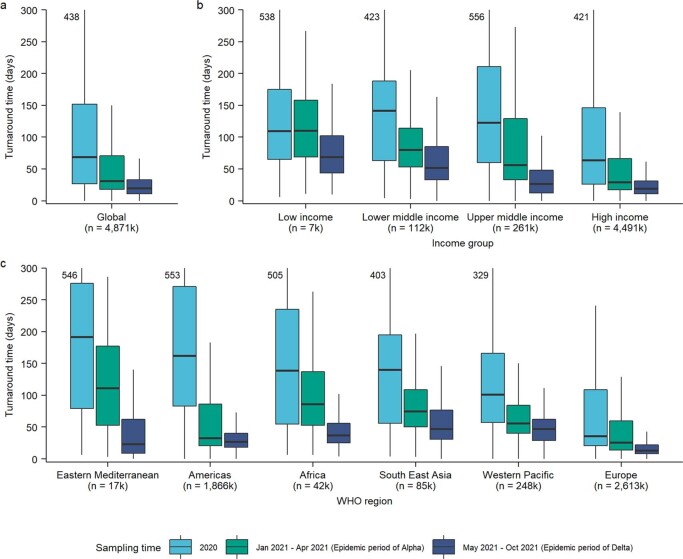
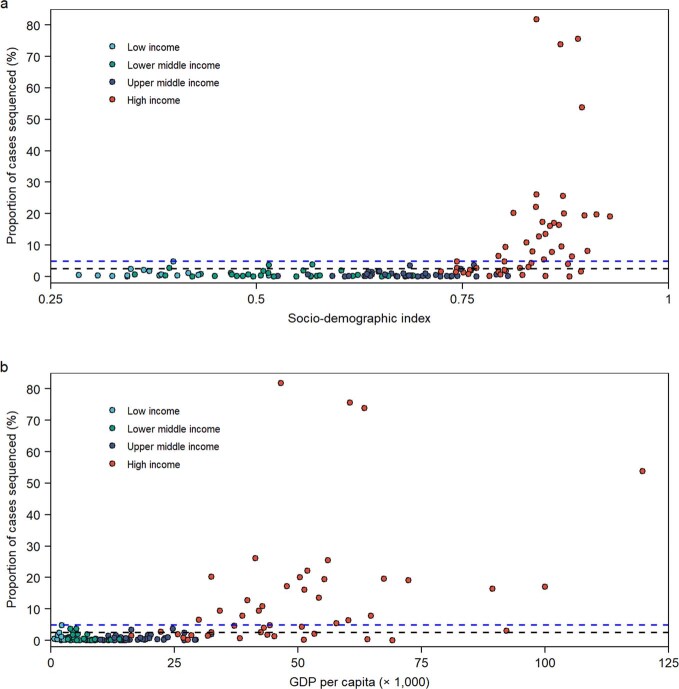
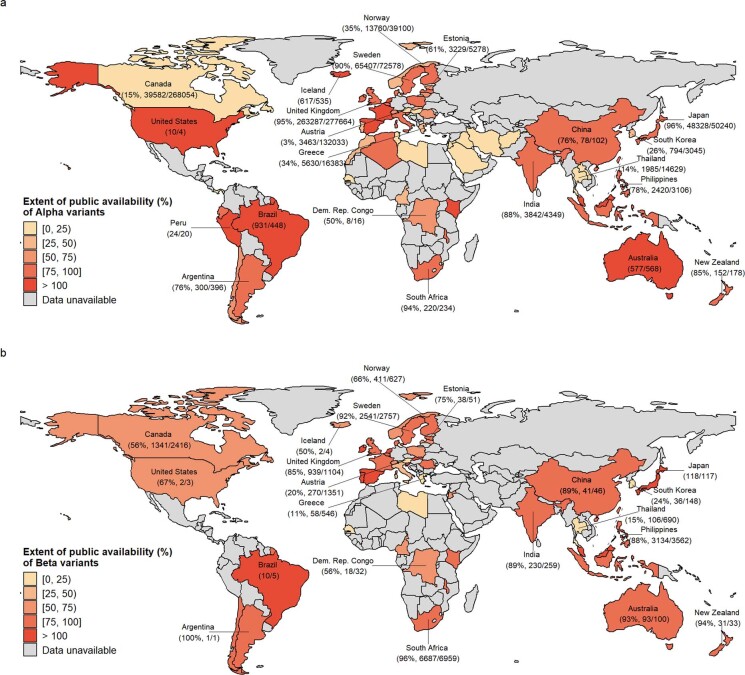
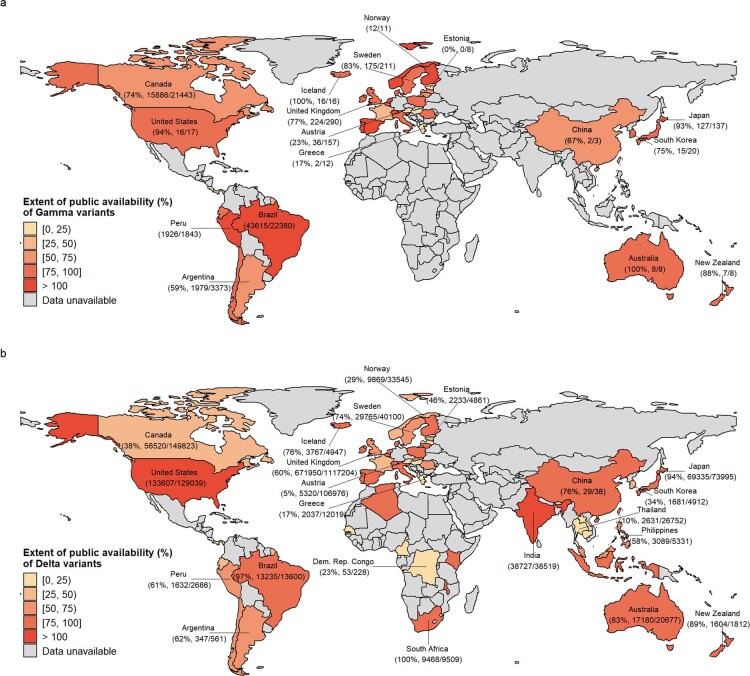
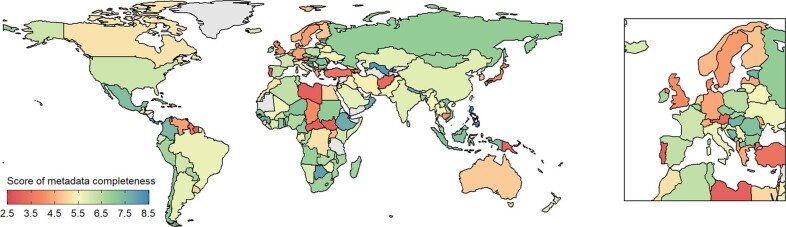
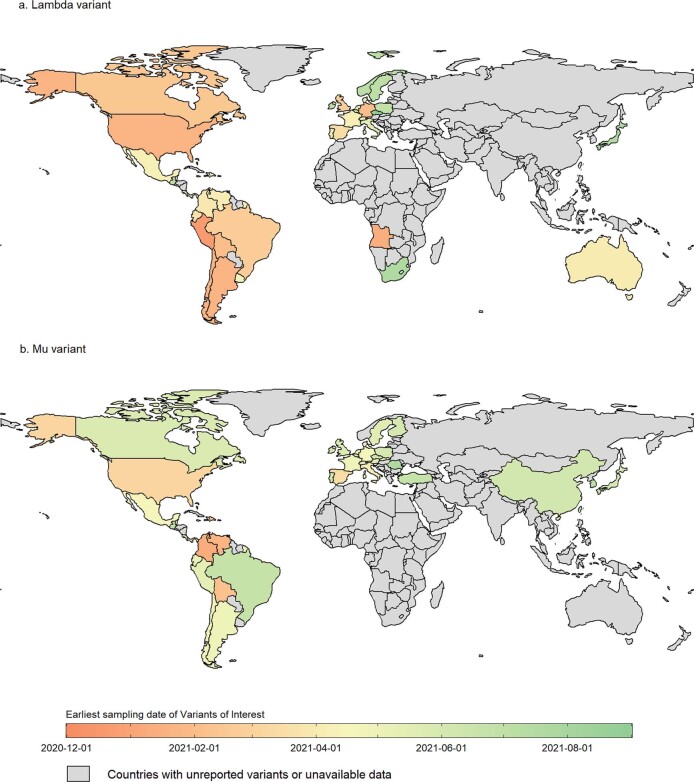
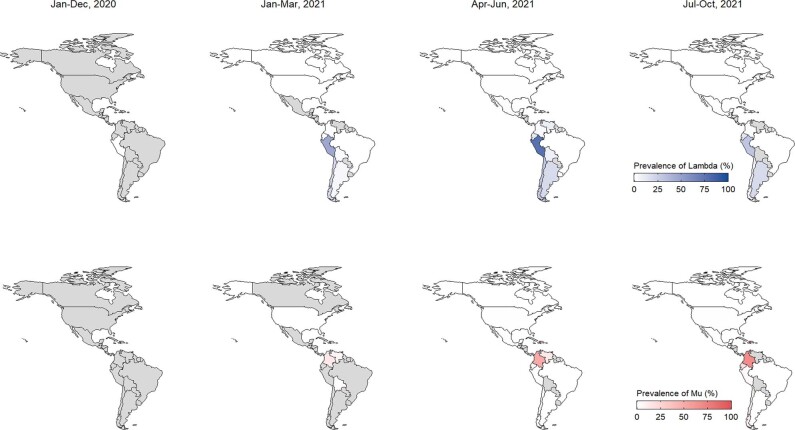
Genomic surveillance has shaped our understanding of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants. We performed a global landscape analysis on SARS-CoV-2 genomic surveillance and genomic data using a collection of country-specific data. Here, we characterize increasing circulation of the Alpha variant in early 2021, subsequently replaced by the Delta variant around May 2021. SARS-CoV-2 genomic surveillance and sequencing availability varied markedly across countries, with 45 countries performing a high level of routine genomic surveillance and 96 countries with a high availability of SARS-CoV-2 sequencing. We also observed a marked heterogeneity of sequencing percentage, sequencing technologies, turnaround time and completeness of released metadata across regions and income groups. A total of 37% of countries with explicit reporting on variants shared less than half of their sequences of variants of concern (VOCs) in public repositories. Our findings indicate an urgent need to increase timely and full sharing of sequences, the standardization of metadata files and support for countries with limited sequencing and bioinformatics capacity.
© 2022. The Author(s).
Conflict of interest statement
H.Y. has received research funding from Sanofi Pasteur, Shanghai Roche Pharmaceutical Company, and SINOVAC Biotech Ltd. None of those research funding is related to this work. All other authors report no competing interests.
Figures
Update of
-
Global landscape of SARS-CoV-2 genomic surveillance, public availability extent of genomic data, and epidemic shaped by variants.Res Sq [Preprint]. 2021 Sep 29:rs.3.rs-927070. doi: 10.21203/rs.3.rs-927070/v1. Res Sq. 2021. Update in: Nat Genet. 2022 Apr;54(4):499-507. doi: 10.1038/s41588-022-01033-y. PMID: 34611660 Free PMC article. Updated. Preprint.
References
-
- World Health Organization. Tracking SARS-CoV-2 variants. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (2021).
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
