

Networks and Graphs Discovery in Metabolomics Data Analysis and Interpretation
- PMID: 35350714
- PMCID: PMC8957799
- DOI: 10.3389/fmolb.2022.841373
Networks and Graphs Discovery in Metabolomics Data Analysis and Interpretation
Abstract
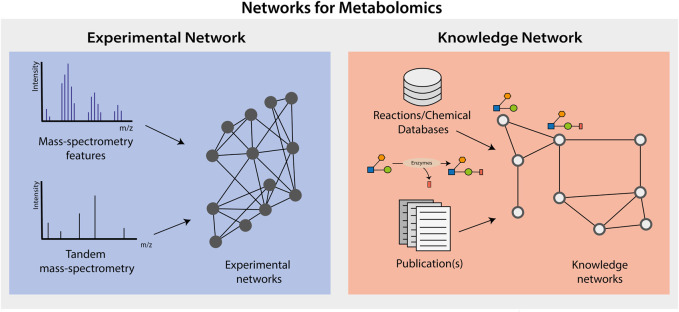
Both targeted and untargeted mass spectrometry-based metabolomics approaches are used to understand the metabolic processes taking place in various organisms, from prokaryotes, plants, fungi to animals and humans. Untargeted approaches allow to detect as many metabolites as possible at once, identify unexpected metabolic changes, and characterize novel metabolites in biological samples. However, the identification of metabolites and the biological interpretation of such large and complex datasets remain challenging. One approach to address these challenges is considering that metabolites are connected through informative relationships. Such relationships can be formalized as networks, where the nodes correspond to the metabolites or features (when there is no or only partial identification), and edges connect nodes if the corresponding metabolites are related. Several networks can be built from a single dataset (or a list of metabolites), where each network represents different relationships, such as statistical (correlated metabolites), biochemical (known or putative substrates and products of reactions), or chemical (structural similarities, ontological relations). Once these networks are built, they can subsequently be mined using algorithms from network (or graph) theory to gain insights into metabolism. For instance, we can connect metabolites based on prior knowledge on enzymatic reactions, then provide suggestions for potential metabolite identifications, or detect clusters of co-regulated metabolites. In this review, we first aim at settling a nomenclature and formalism to avoid confusion when referring to different networks used in the field of metabolomics. Then, we present the state of the art of network-based methods for mass spectrometry-based metabolomics data analysis, as well as future developments expected in this area. We cover the use of networks applications using biochemical reactions, mass spectrometry features, chemical structural similarities, and correlations between metabolites. We also describe the application of knowledge networks such as metabolic reaction networks. Finally, we discuss the possibility of combining different networks to analyze and interpret them simultaneously.
Keywords: experimental network; graph-based analysis; knowledge network; metabolic network; metabolism; systems biology; untargeted metabolomics.
Copyright © 2022 Amara, Frainay, Jourdan, Naake, Neumann, Novoa-del-Toro, Salek, Salzer, Scharfenberg and Witting.
Conflict of interest statement
Author RS is employed by Bruker BioSpin GmbH. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures



References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources