

Killing the competition: a theoretical framework for liver-stage malaria
- PMID: 35350863
- PMCID: PMC8965401
- DOI: 10.1098/rsob.210341
Killing the competition: a theoretical framework for liver-stage malaria
Abstract
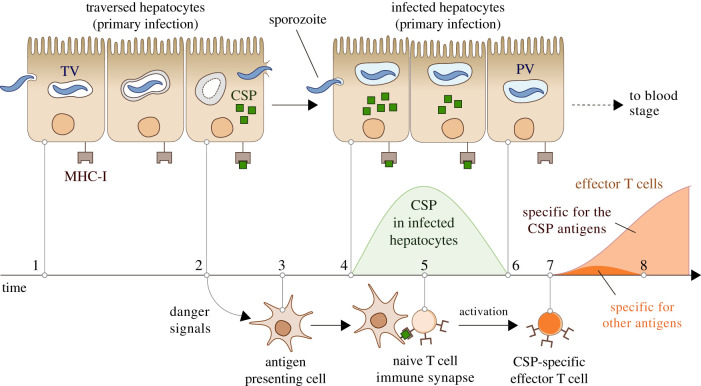
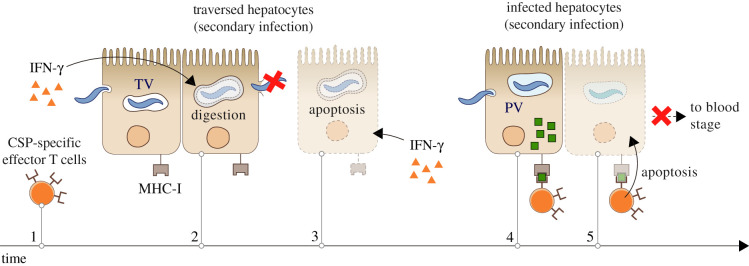
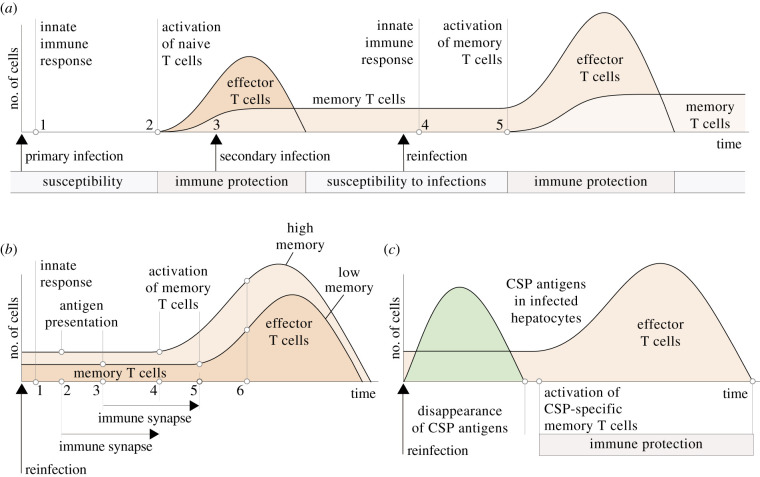
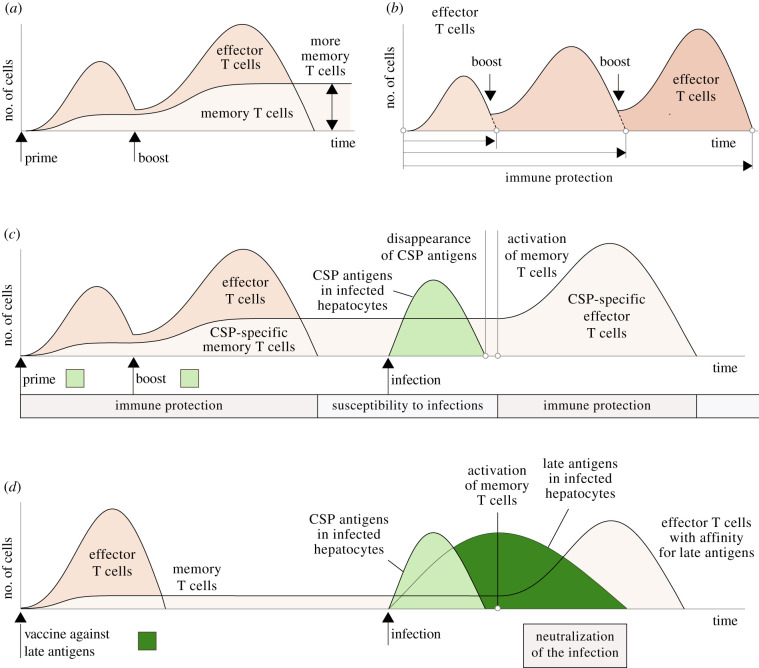
The first stage of malaria infections takes place inside the host's hepatocytes. Remarkably, Plasmodium parasites do not infect hepatocytes immediately after reaching the liver. Instead, they migrate through several hepatocytes before infecting their definitive host cells, thus increasing their chances of immune destruction. Considering that malaria can proceed normally without cell traversal, this is indeed a puzzling behaviour. In fact, the role of hepatocyte traversal remains unknown to date, implying that the current understanding of malaria is incomplete. In this work, we hypothesize that the parasites traverse hepatocytes to actively trigger an immune response in the host. This behaviour would be part of a strategy of superinfection exclusion aimed to reduce intraspecific competition during the blood stage of the infection. Based on this hypothesis, we formulate a comprehensive theory of liver-stage malaria that integrates all the available knowledge about the infection. The interest of this new paradigm is not merely theoretical. It highlights major issues in the current empirical approach to the study of Plasmodium and suggests new strategies to fight malaria.
Keywords: Plasmodium; antimalarial vaccines; concomitant immunity; liver-stage malaria; superinfection exclusion.
Conflict of interest statement
We declare we have no competing interests.
Figures
Similar articles
-
Plasmodium sporozoites on the move: Switching from cell traversal to productive invasion of hepatocytes.Mol Microbiol. 2021 May;115(5):870-881. doi: 10.1111/mmi.14645. Epub 2020 Dec 5. Mol Microbiol. 2021. PMID: 33191548 Free PMC article. Review.
-
Monocyte-Derived CD11c+ Cells Acquire Plasmodium from Hepatocytes to Prime CD8 T Cell Immunity to Liver-Stage Malaria.Cell Host Microbe. 2019 Apr 10;25(4):565-577.e6. doi: 10.1016/j.chom.2019.02.014. Epub 2019 Mar 21. Cell Host Microbe. 2019. PMID: 30905437 Free PMC article.
-
Sneaking in through the back entrance: the biology of malaria liver stages.Trends Parasitol. 2004 Sep;20(9):417-24. doi: 10.1016/j.pt.2004.07.007. Trends Parasitol. 2004. PMID: 15324732 Review.
-
Susceptibility to Plasmodium liver stage infection is altered by hepatocyte polyploidy.Cell Microbiol. 2014 May;16(5):784-95. doi: 10.1111/cmi.12282. Epub 2014 Mar 28. Cell Microbiol. 2014. PMID: 24612025 Free PMC article.
-
Gene disruption of Plasmodium falciparum p52 results in attenuation of malaria liver stage development in cultured primary human hepatocytes.PLoS One. 2008;3(10):e3549. doi: 10.1371/journal.pone.0003549. Epub 2008 Oct 28. PLoS One. 2008. PMID: 18958160 Free PMC article.
Cited by
-
SIRI+Q model with a limited capacity of isolation.Theory Biosci. 2025 Jun;144(2):121-144. doi: 10.1007/s12064-025-00437-8. Epub 2025 Mar 29. Theory Biosci. 2025. PMID: 40158025 Free PMC article.
-
What Influence Could the Acceptance of Visitors Cause on the Epidemic Dynamics of a Reinfectious Disease?: A Mathematical Model.Acta Biotheor. 2024 Feb 25;72(1):3. doi: 10.1007/s10441-024-09478-w. Acta Biotheor. 2024. PMID: 38402514 Free PMC article.
