

Covalent Proximity Scanning of a Distal Cysteine to Target PI3Kα
- PMID: 35353516
- PMCID: PMC9011356
- DOI: 10.1021/jacs.1c13568
Covalent Proximity Scanning of a Distal Cysteine to Target PI3Kα
Abstract
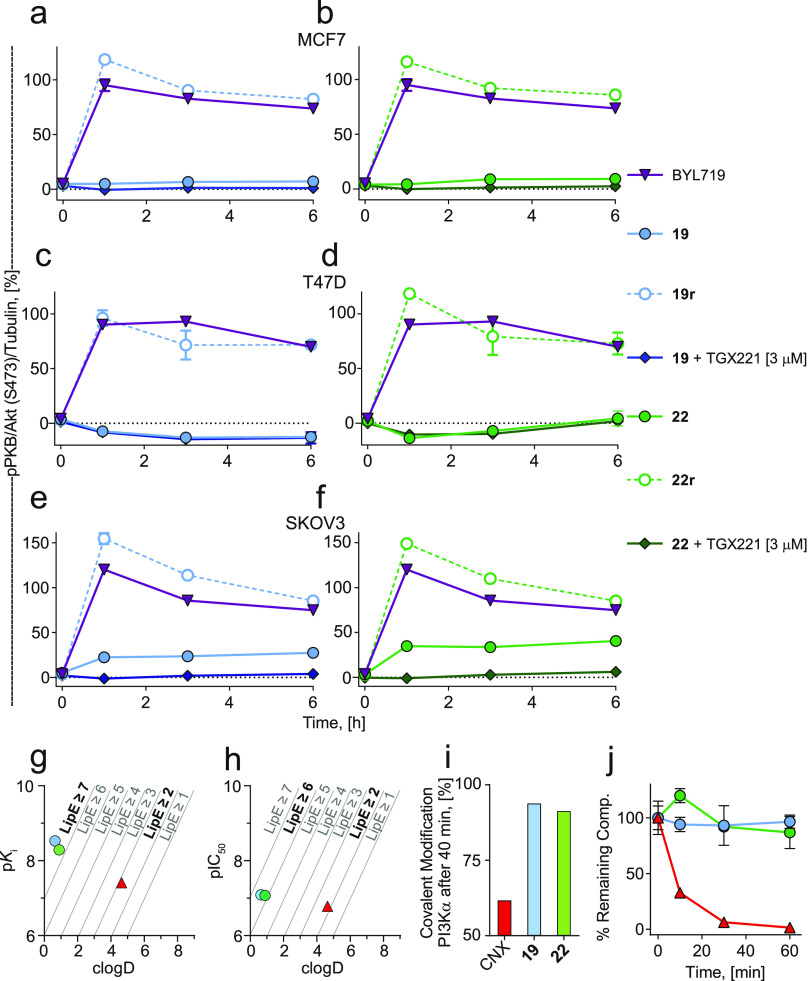
Covalent protein kinase inhibitors exploit currently noncatalytic cysteines in the adenosine 5'-triphosphate (ATP)-binding site via electrophiles directly appended to a reversible-inhibitor scaffold. Here, we delineate a path to target solvent-exposed cysteines at a distance >10 Å from an ATP-site-directed core module and produce potent covalent phosphoinositide 3-kinase α (PI3Kα) inhibitors. First, reactive warheads are used to reach out to Cys862 on PI3Kα, and second, enones are replaced with druglike warheads while linkers are optimized. The systematic investigation of intrinsic warhead reactivity (kchem), rate of covalent bond formation and proximity (kinact and reaction space volume Vr), and integration of structure data, kinetic and structural modeling, led to the guided identification of high-quality, covalent chemical probes. A novel stochastic approach provided direct access to the calculation of overall reaction rates as a function of kchem, kinact, Ki, and Vr, which was validated with compounds with varied linker lengths. X-ray crystallography, protein mass spectrometry (MS), and NanoBRET assays confirmed covalent bond formation of the acrylamide warhead and Cys862. In rat liver microsomes, compounds 19 and 22 outperformed the rapidly metabolized CNX-1351, the only known PI3Kα irreversible inhibitor. Washout experiments in cancer cell lines with mutated, constitutively activated PI3Kα showed a long-lasting inhibition of PI3Kα. In SKOV3 cells, compounds 19 and 22 revealed PI3Kβ-dependent signaling, which was sensitive to TGX221. Compounds 19 and 22 thus qualify as specific chemical probes to explore PI3Kα-selective signaling branches. The proposed approach is generally suited to develop covalent tools targeting distal, unexplored Cys residues in biologically active enzymes.
Conflict of interest statement
The authors declare no competing financial interest.
Figures






References
-
- Miller V. A.; Hirsh V.; Cadranel J.; Chen Y.-M.; Park K.; Kim S.-W.; Zhou C.; Su W.-C.; Wang M.; Sun Y.; Heo D. S.; Crino L.; Tan E.-H.; Chao T.-Y.; Shahidi M.; Cong X. J.; Lorence R. M.; Yang J. C.-H. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012, 13, 528–538. 10.1016/S1470-2045(12)70087-6. - DOI - PubMed
-
- Pan Z.; Scheerens H.; Li S.-J.; Schultz B. E.; Sprengeler P. A.; Burrill L. C.; Mendonca R. V.; Sweeney M. D.; Scott K. C. K.; Grothaus P. G.; Jeffery D. A.; Spoerke J. M.; Honigberg L. A.; Young P. R.; Dalrymple S. A.; Palmer J. T. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem 2007, 2, 58–61. 10.1002/cmdc.200600221. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
