

A PI3Kγ mimetic peptide triggers CFTR gating, bronchodilation, and reduced inflammation in obstructive airway diseases
- PMID: 35353541
- PMCID: PMC9869178
- DOI: 10.1126/scitranslmed.abl6328
A PI3Kγ mimetic peptide triggers CFTR gating, bronchodilation, and reduced inflammation in obstructive airway diseases
Abstract
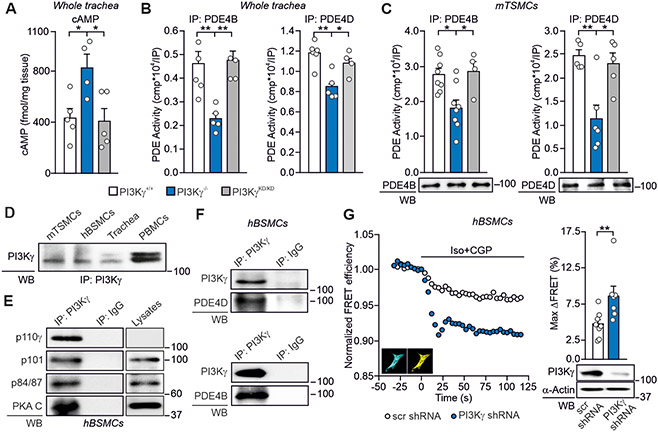
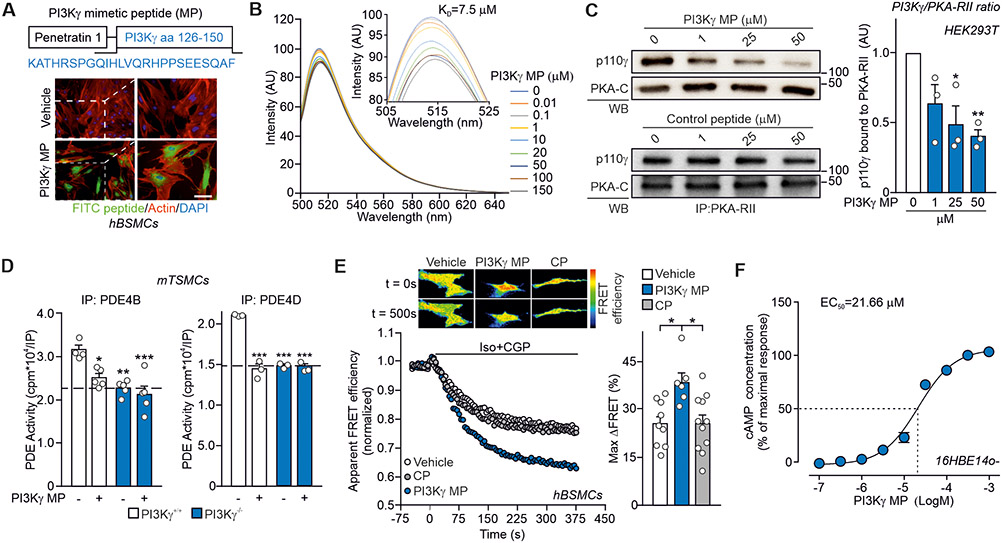
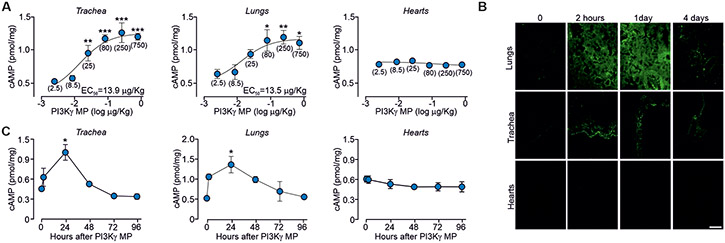
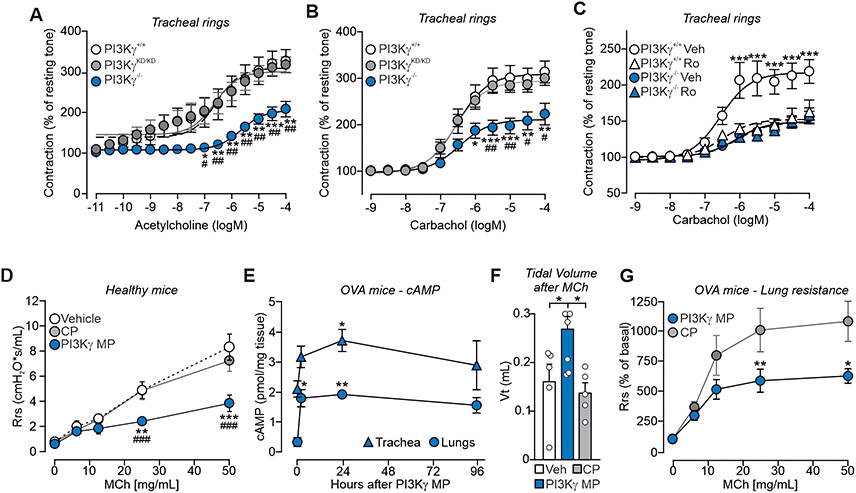
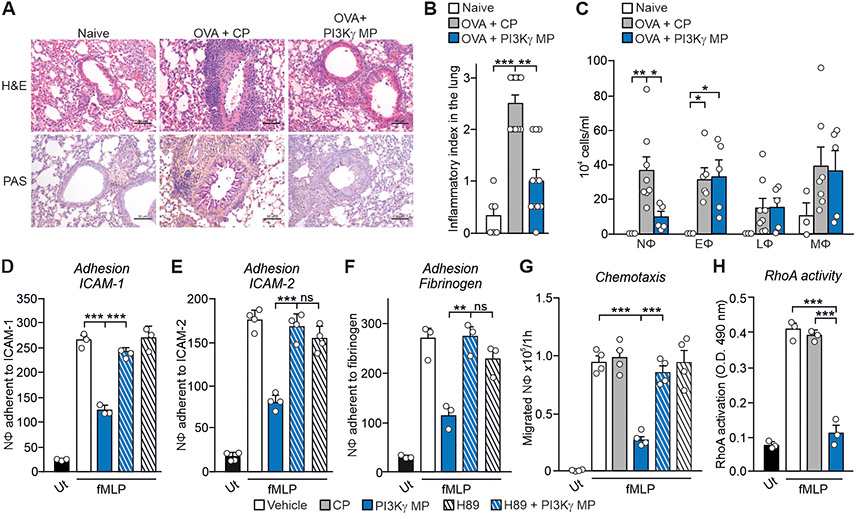
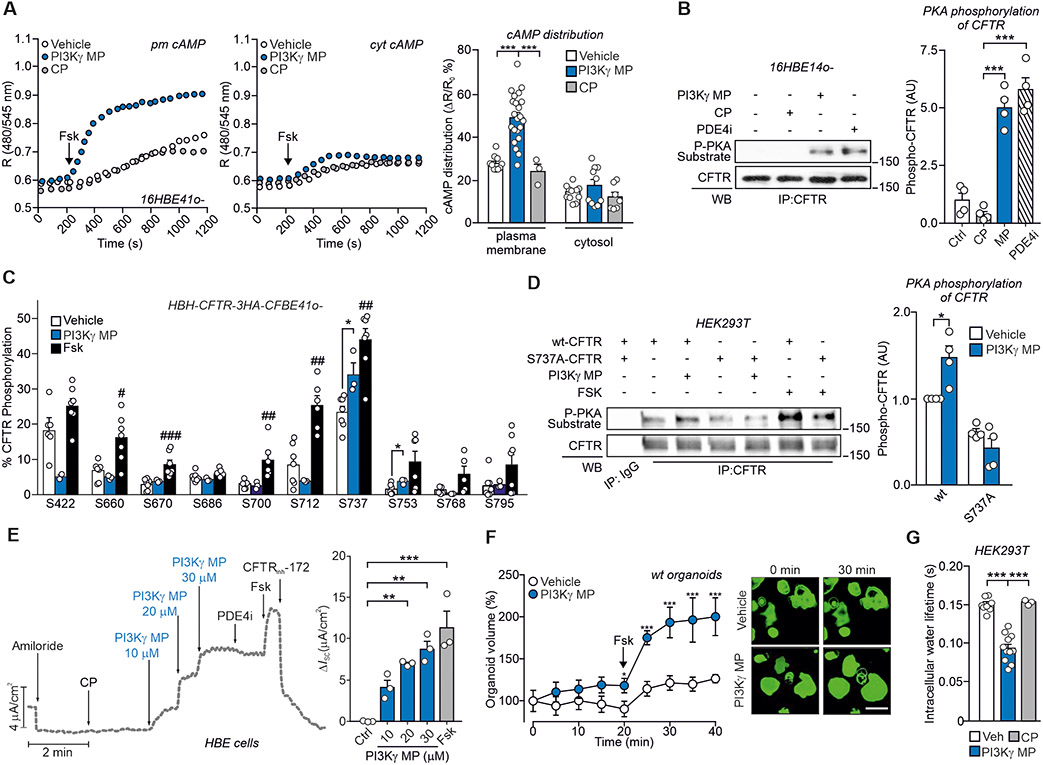
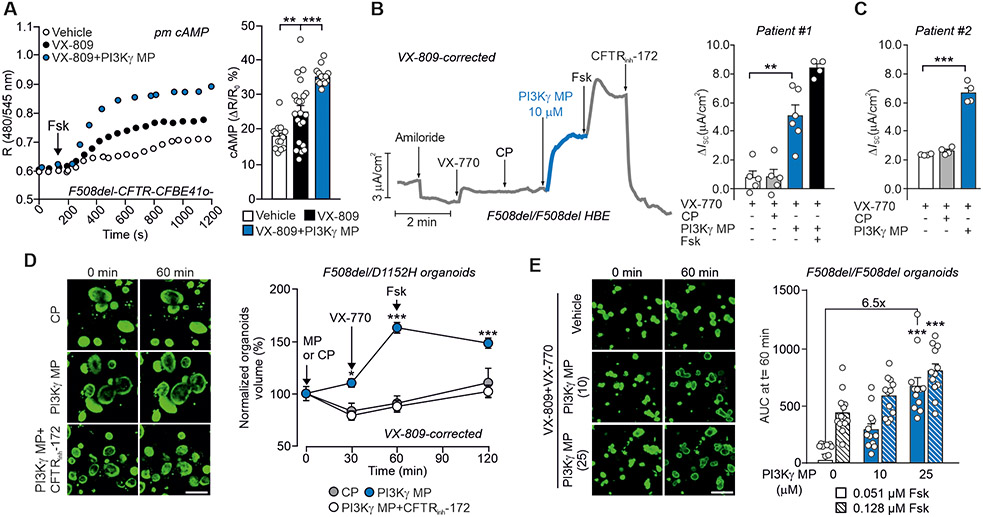
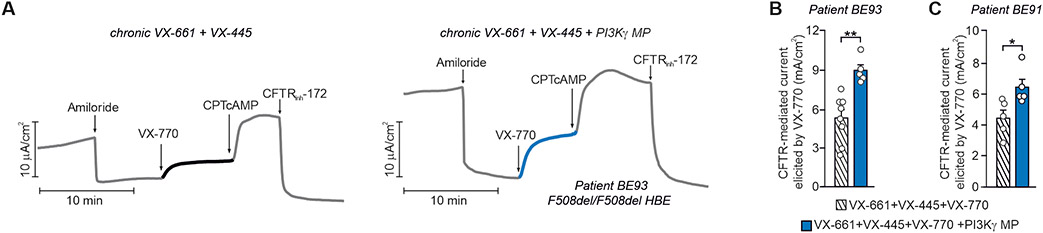
Cyclic adenosine 3',5'-monophosphate (cAMP)-elevating agents, such as β2-adrenergic receptor (β2-AR) agonists and phosphodiesterase (PDE) inhibitors, remain a mainstay in the treatment of obstructive respiratory diseases, conditions characterized by airway constriction, inflammation, and mucus hypersecretion. However, their clinical use is limited by unwanted side effects because of unrestricted cAMP elevation in the airways and in distant organs. Here, we identified the A-kinase anchoring protein phosphoinositide 3-kinase γ (PI3Kγ) as a critical regulator of a discrete cAMP signaling microdomain activated by β2-ARs in airway structural and inflammatory cells. Displacement of the PI3Kγ-anchored pool of protein kinase A (PKA) by an inhaled, cell-permeable, PI3Kγ mimetic peptide (PI3Kγ MP) inhibited a pool of subcortical PDE4B and PDE4D and safely increased cAMP in the lungs, leading to airway smooth muscle relaxation and reduced neutrophil infiltration in a murine model of asthma. In human bronchial epithelial cells, PI3Kγ MP induced unexpected cAMP and PKA elevations restricted to the vicinity of the cystic fibrosis transmembrane conductance regulator (CFTR), the ion channel controlling mucus hydration that is mutated in cystic fibrosis (CF). PI3Kγ MP promoted the phosphorylation of wild-type CFTR on serine-737, triggering channel gating, and rescued the function of F508del-CFTR, the most prevalent CF mutant, by enhancing the effects of existing CFTR modulators. These results unveil PI3Kγ as the regulator of a β2-AR/cAMP microdomain central to smooth muscle contraction, immune cell activation, and epithelial fluid secretion in the airways, suggesting the use of a PI3Kγ MP for compartment-restricted, therapeutic cAMP elevation in chronic obstructive respiratory diseases.
Figures
References
-
- Celli BR, Wedzicha JA, Update on Clinical Aspects of Chronic Obstructive Pulmonary Disease. N Engl J Med 381, 1257–1266 (2019). - PubMed
-
- Shteinberg M, Haq IJ, Polineni D, Davies JC, Cystic fibrosis. Lancet 397, 2195–2211 (2021). - PubMed
-
- Barnes PJ, New drugs for asthma. Nat Rev Drug Discov 3, 831–844 (2004). - PubMed
-
- Vijftigschild LA, Berkers G, Dekkers JF, Zomer-van Ommen DD, Matthes E, Kruisselbrink E, Vonk A, Hensen CE, Heida-Michel S, Geerdink M, Janssens HM, van de Graaf EA, Bronsveld I, de Winter-de Groot KM, Majoor CJ, Heijerman HG, de Jonge HR, Hanrahan JW, van der Ent CK, Beekman JM, beta2-Adrenergic receptor agonists activate CFTR in intestinal organoids and subjects with cystic fibrosis. Eur Respir J 48, 768–779 (2016). - PubMed
-
- Dunican EM, Elicker BM, Henry T, Gierada DS, Schiebler ML, Anderson W, Barjaktarevic I, Barr RG, Bleecker ER, Boucher RC, Bowler R, Christenson SA, Comellas A, Cooper CB, Couper D, Criner GJ, Dransfield M, Doerschuk CM, Drummond MB, Hansel NN, Han MK, Hastie AT, Hoffman EA, Krishnan JA, Lazarus SC, Martinez FJ, McCulloch CE, O'Neal WK, Ortega VE, Paine R 3rd, Peters S, Schroeder JD, Woodruff PG, Fahy JV, Mucus Plugs and Emphysema in the Pathophysiology of Airflow Obstruction and Hypoxemia in Smokers. Am J Respir Crit Care Med 203, 957–968 (2021). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
