

Small-molecule targeted therapies induce dependence on DNA double-strand break repair in residual tumor cells
- PMID: 35353542
- PMCID: PMC9516479
- DOI: 10.1126/scitranslmed.abc7480
Small-molecule targeted therapies induce dependence on DNA double-strand break repair in residual tumor cells
Abstract
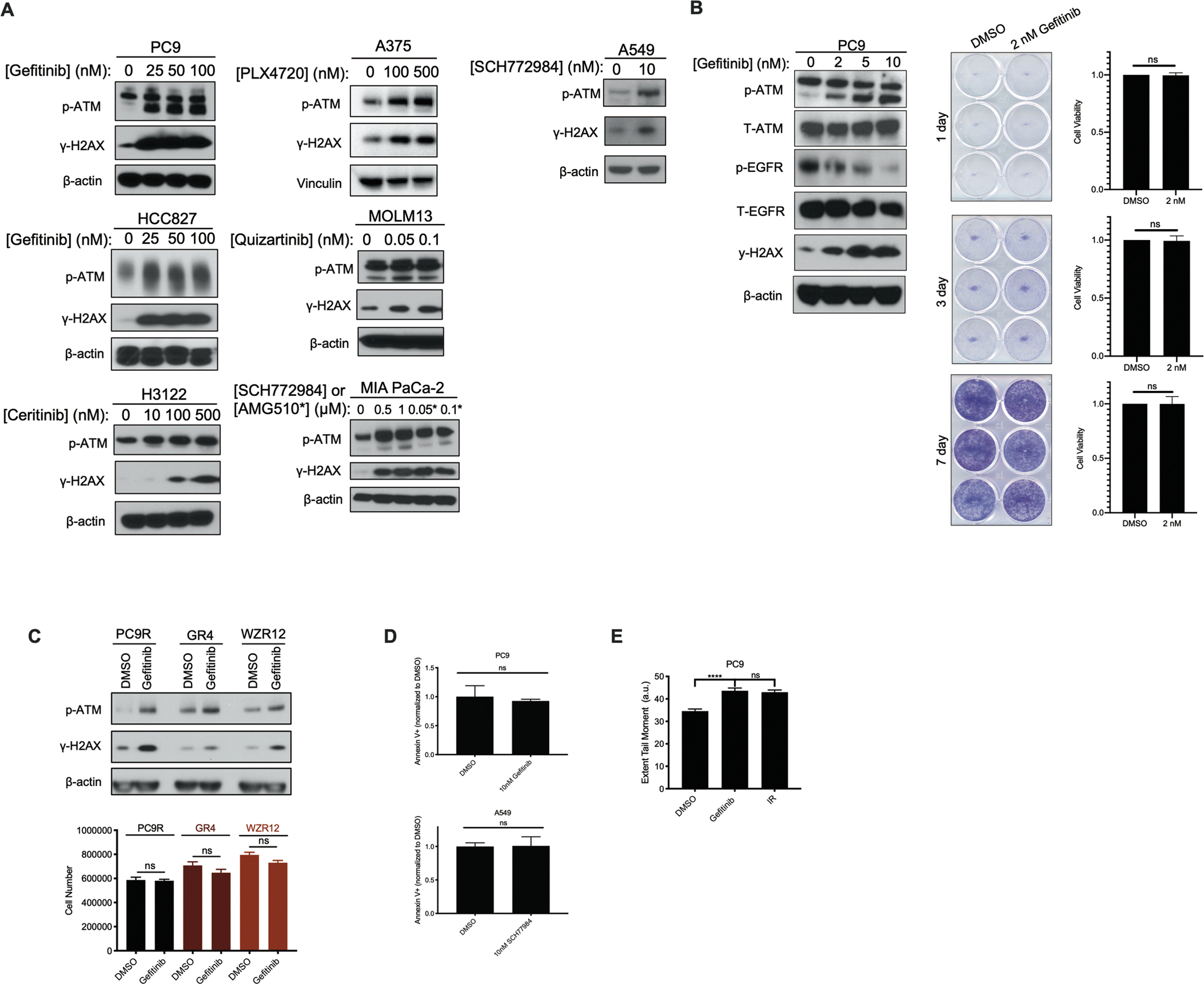
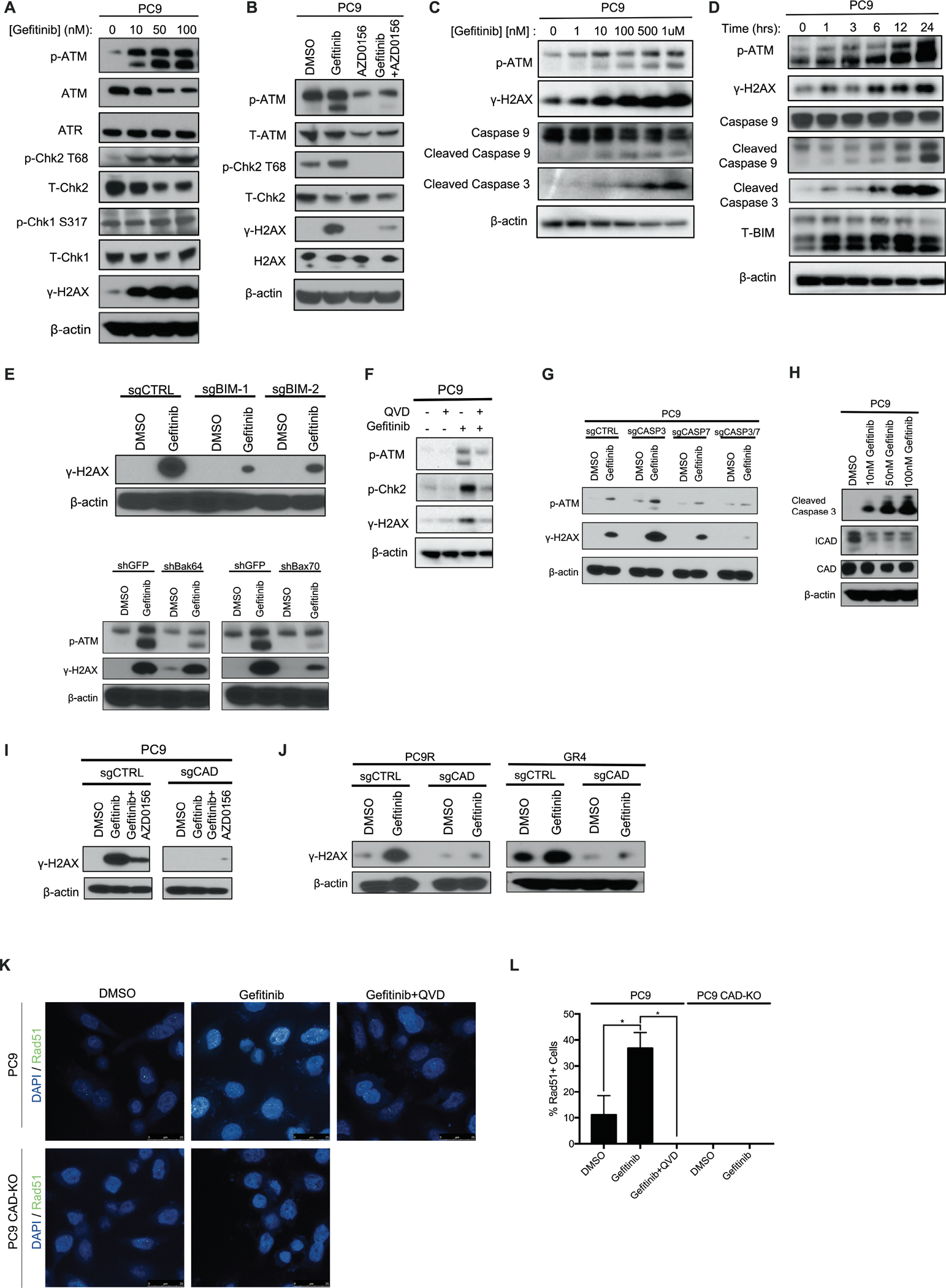
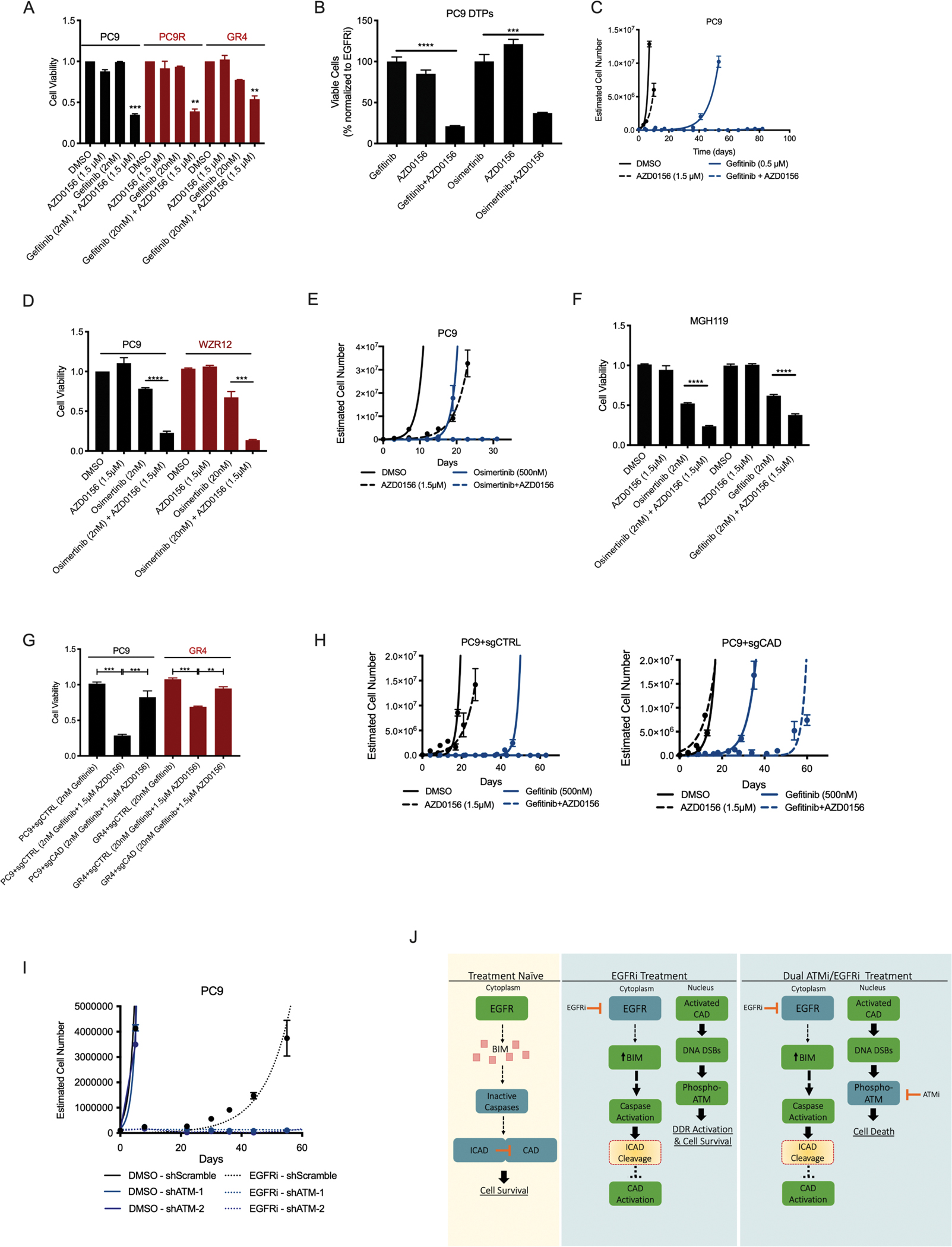
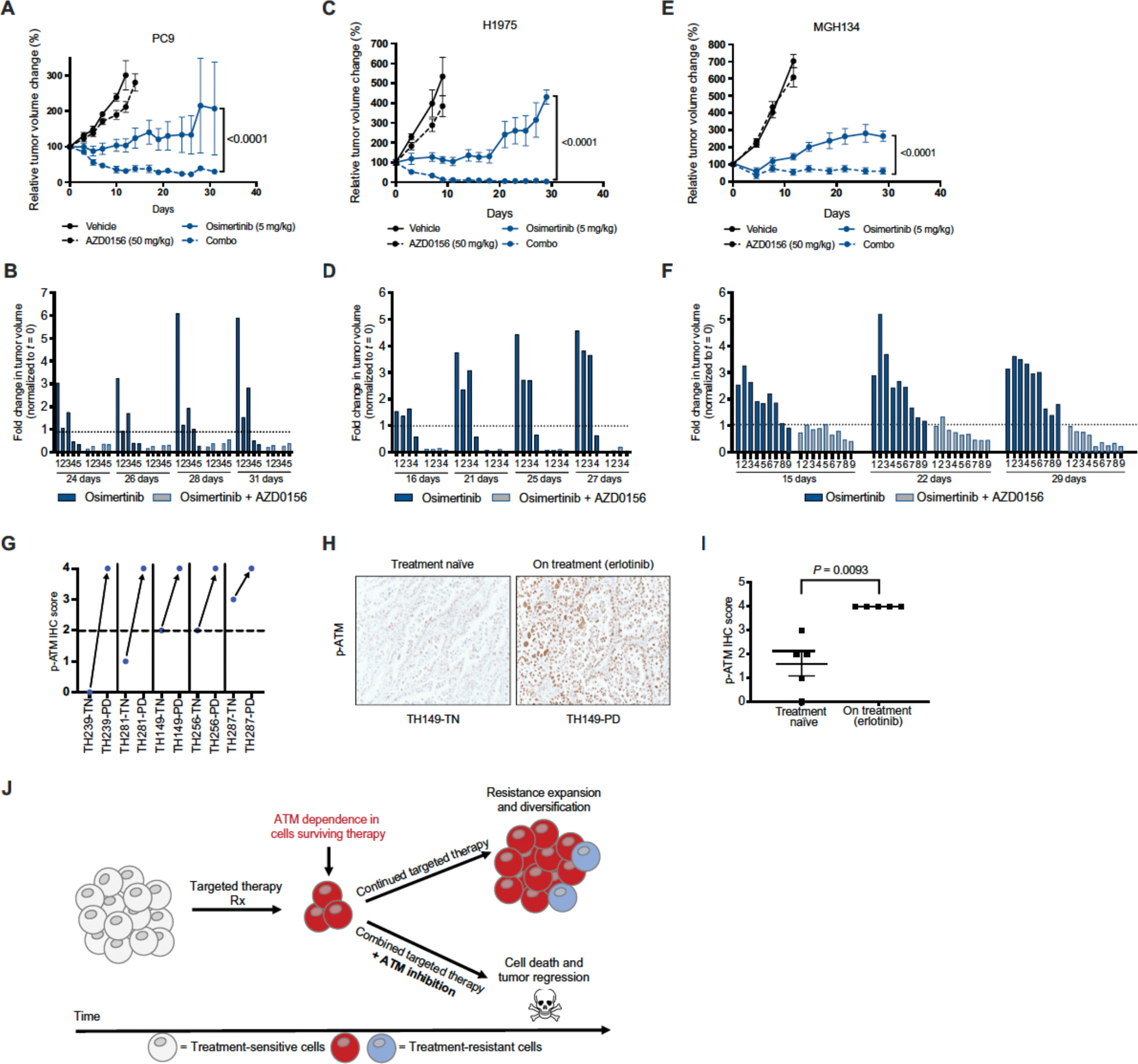
Residual cancer cells that survive drug treatments with targeted therapies act as a reservoir from which eventual resistant disease emerges. Although there is great interest in therapeutically targeting residual cells, efforts are hampered by our limited knowledge of the vulnerabilities existing in this cell state. Here, we report that diverse oncogene-targeted therapies, including inhibitors of epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), KRAS, and BRAF, induce DNA double-strand breaks and, consequently, ataxia-telangiectasia mutated (ATM)-dependent DNA repair in oncogene-matched residual tumor cells. This DNA damage response, observed in cell lines, mouse xenograft models, and human patients, is driven by a pathway involving the activation of caspases 3 and 7 and the downstream caspase-activated deoxyribonuclease (CAD). CAD is, in turn, activated through caspase-mediated degradation of its endogenous inhibitor, ICAD. In models of EGFR mutant non-small cell lung cancer (NSCLC), tumor cells that survive treatment with small-molecule EGFR-targeted therapies are thus synthetically dependent on ATM, and combined treatment with an ATM kinase inhibitor eradicates these cells in vivo. This led to more penetrant and durable responses in EGFR mutant NSCLC mouse xenograft models, including those derived from both established cell lines and patient tumors. Last, we found that rare patients with EGFR mutant NSCLC harboring co-occurring, loss-of-function mutations in ATM exhibit extended progression-free survival on first generation EGFR inhibitor therapy relative to patients with EGFR mutant NSCLC lacking deleterious ATM mutations. Together, these findings establish a rationale for the mechanism-based integration of ATM inhibitors alongside existing targeted therapies.
Conflict of interest statement
Competing Interests
K.C.W. is a co-founder and consultant for Element Genomics, Tavros Therapeutics, and Celldom. A.N.H. has served as a consultant for Nuvalent, Inc., and receives research funding from Pfizer, Relay Therapeutics, Roche/Genentech, Eli Lilly, Blueprint Medicines and Amgen. T.G.B. is an advisor to Novartis, Astrazeneca, Revolution Medicines, Array/Pfizer, Springworks, Strategia, Relay, Jazz, Rain, EcoR1 and receives research funding from Novartis and Revolution Medicines and Strategia. C.M.B has performed consulting work for Revolution Medicines, Blueprints Medicine, Amgen, Bayer, and Foundation Medicine. H.A.Y. has performed consulting work for Daiichi, Janssen, Blueprint Medicine and AstraZeneca. C.G. is a scientific advisor for SafineAI, LLC. C.E.M is an employee of Genentech Inc, has performed unpaid consulting for Eli Lilly and Loxo, has received honoraria from Genentech, Astra Zeneca, Takeda, Novartis and Guardant Health, and receives/received research funding from Novartis and Revolution Medicines.
Figures
References
-
- Haber DA, Gray NS, Baselga J, The evolving war on cancer. Cell 145, 19–24 (2011). - PubMed
-
- Chae YK, Pan AP, Davis AA, Patel SP, Carneiro BA, Kurzrock R, Giles FJ, Path toward Precision Oncology: Review of Targeted Therapy Studies and Tools to Aid in Defining “Actionability” of a Molecular Lesion and Patient Management Support. Mol. Cancer Ther. 16, 2645–2655 (2017). - PubMed
-
- Amirouchene-Angelozzi N, Swanton C, Bardelli A, Tumor Evolution as a Therapeutic Target. Cancer Discov. 7, 805–817 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 CA217882/CA/NCI NIH HHS/United States
- F32 CA206234/CA/NCI NIH HHS/United States
- U54 CA224081/CA/NCI NIH HHS/United States
- R01 CA211052/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- F30 CA220847/CA/NCI NIH HHS/United States
- R01 CA207083/CA/NCI NIH HHS/United States
- T32 GM007171/GM/NIGMS NIH HHS/United States
- R01 CA204302/CA/NCI NIH HHS/United States
- P30 CA014236/CA/NCI NIH HHS/United States
- T32 GM145449/GM/NIGMS NIH HHS/United States
- R01 CA231300/CA/NCI NIH HHS/United States
- R01 CA169338/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
