

CREBBP/EP300 acetyltransferase inhibition disrupts FOXA1-bound enhancers to inhibit the proliferation of ER+ breast cancer cells
- PMID: 35353838
- PMCID: PMC8967035
- DOI: 10.1371/journal.pone.0262378
CREBBP/EP300 acetyltransferase inhibition disrupts FOXA1-bound enhancers to inhibit the proliferation of ER+ breast cancer cells
Abstract
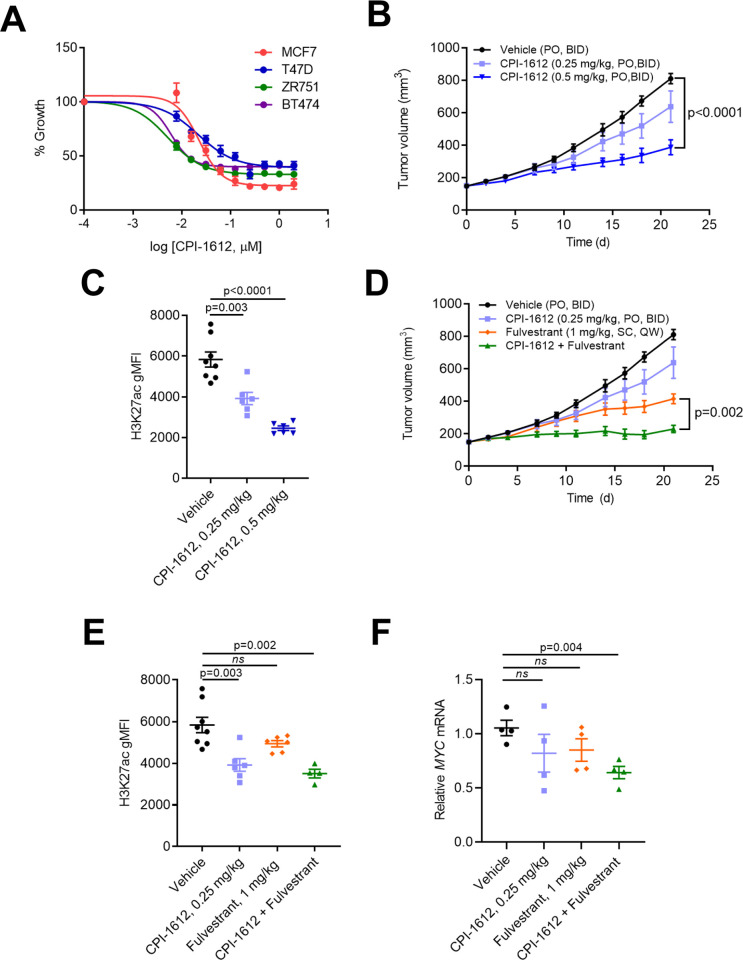
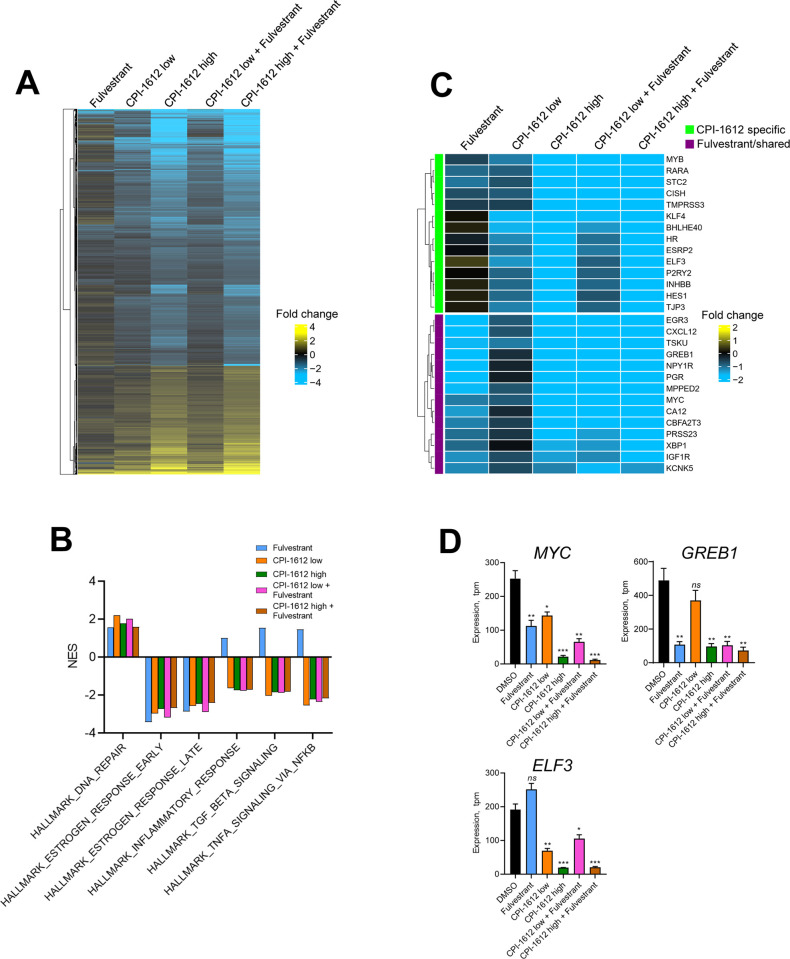
Therapeutic targeting of the estrogen receptor (ER) is a clinically validated approach for estrogen receptor positive breast cancer (ER+ BC), but sustained response is limited by acquired resistance. Targeting the transcriptional coactivators required for estrogen receptor activity represents an alternative approach that is not subject to the same limitations as targeting estrogen receptor itself. In this report we demonstrate that the acetyltransferase activity of coactivator paralogs CREBBP/EP300 represents a promising therapeutic target in ER+ BC. Using the potent and selective inhibitor CPI-1612, we show that CREBBP/EP300 acetyltransferase inhibition potently suppresses in vitro and in vivo growth of breast cancer cell line models and acts in a manner orthogonal to directly targeting ER. CREBBP/EP300 acetyltransferase inhibition suppresses ER-dependent transcription by targeting lineage-specific enhancers defined by the pioneer transcription factor FOXA1. These results validate CREBBP/EP300 acetyltransferase activity as a viable target for clinical development in ER+ breast cancer.
Conflict of interest statement
Authors are current or former employees and stockholders of Constellation Pharmaceuticals, a Morphosys company, which provided funding for this research. Patents have been filed around the chemical series that includes CPI-1612. This work does not relate to any marketed products or products in development. These disclosures do not alter our adherence to PLOS ONE policies on sharing data and materials.
Figures


References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
