

RAS activation induces synthetic lethality of MEK inhibition with mitochondrial oxidative metabolism in acute myeloid leukemia
- PMID: 35354920
- PMCID: PMC9061298
- DOI: 10.1038/s41375-022-01541-0
RAS activation induces synthetic lethality of MEK inhibition with mitochondrial oxidative metabolism in acute myeloid leukemia
Abstract
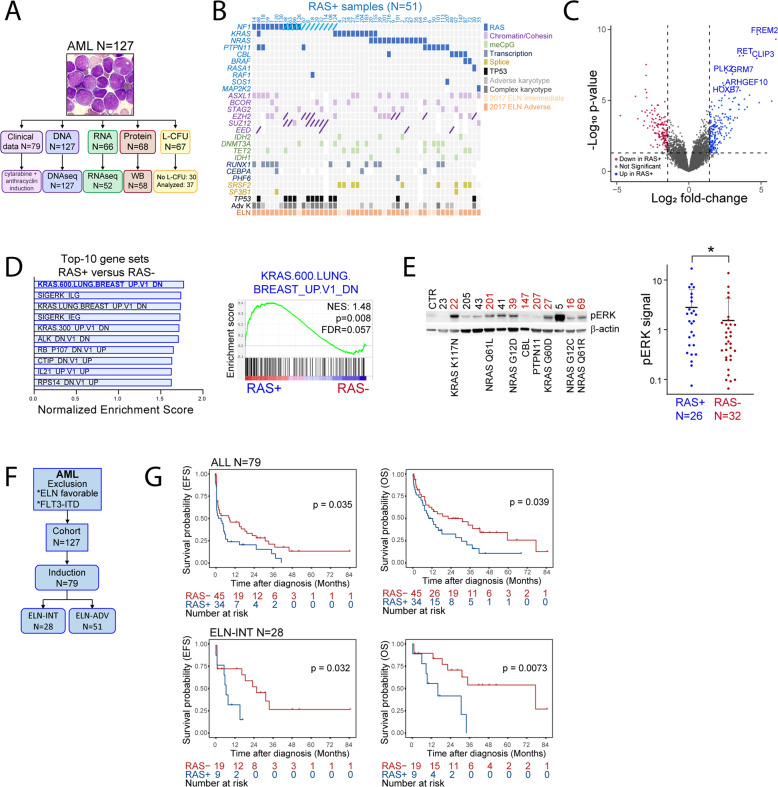
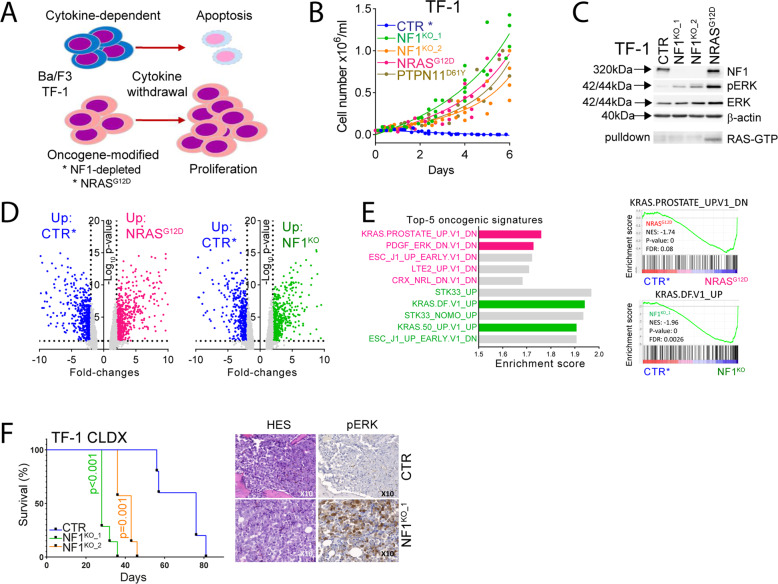
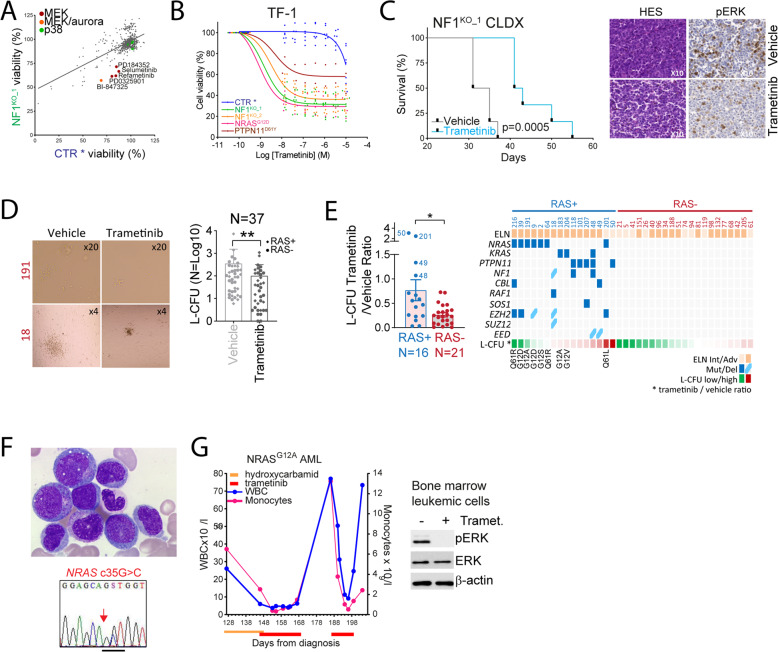
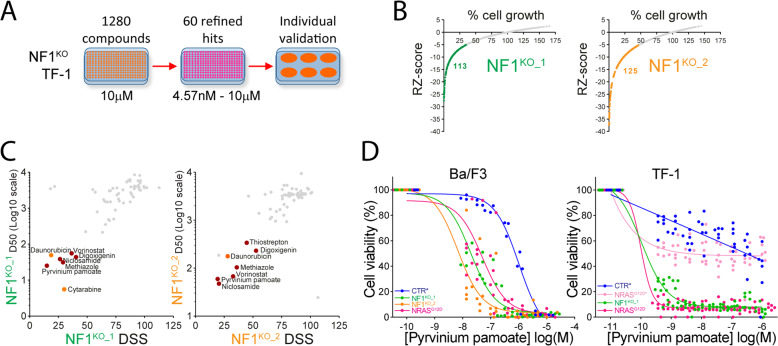
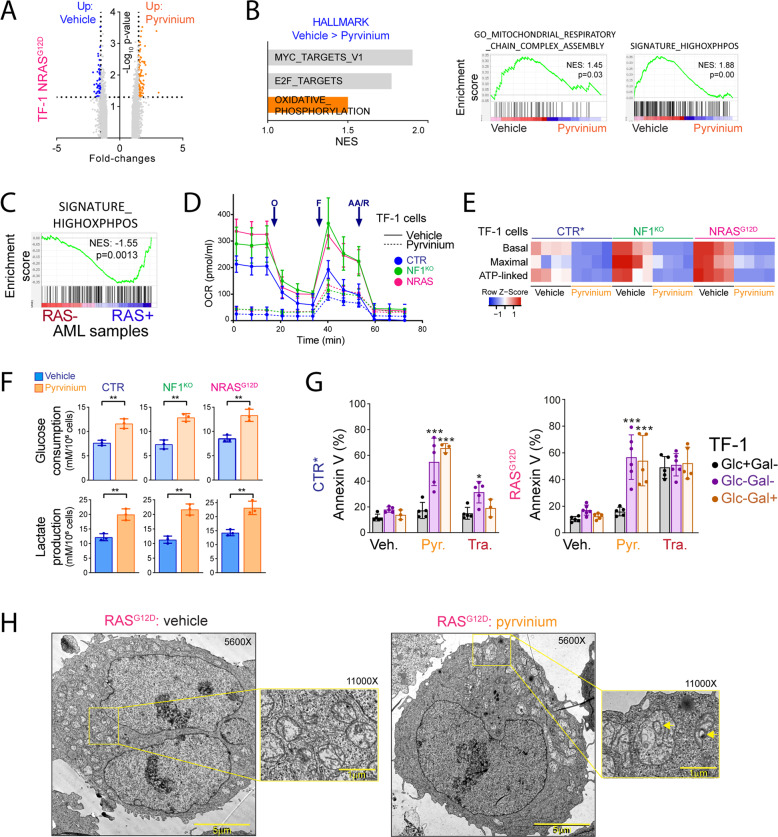
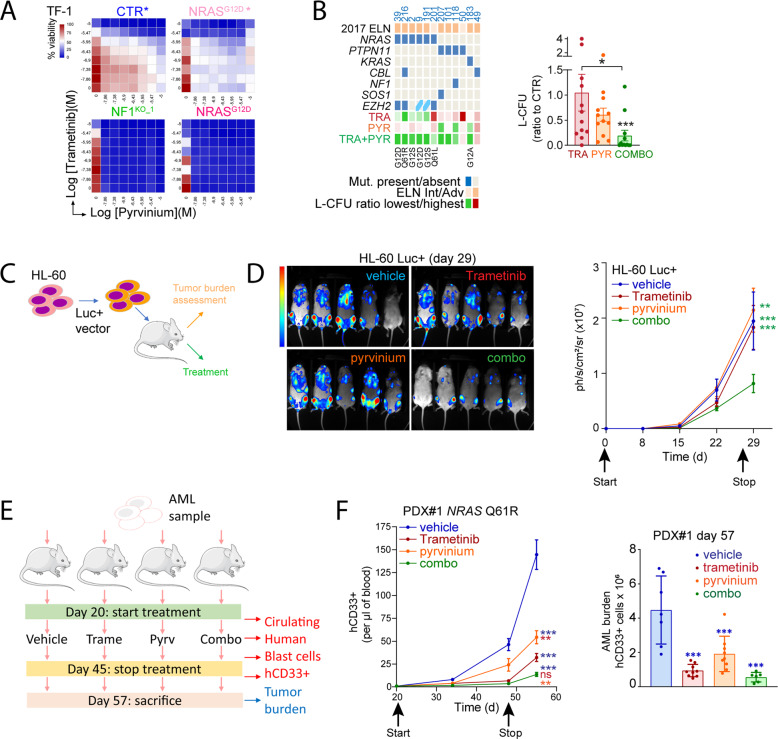
Despite recent advances in acute myeloid leukemia (AML) molecular characterization and targeted therapies, a majority of AML cases still lack therapeutically actionable targets. In 127 AML cases with unmet therapeutic needs, as defined by the exclusion of ELN favorable cases and of FLT3-ITD mutations, we identified 51 (40%) cases with alterations in RAS pathway genes (RAS+, mostly NF1, NRAS, KRAS, and PTPN11 genes). In 79 homogeneously treated AML patients from this cohort, RAS+ status were associated with higher white blood cell count, higher LDH, and reduced survival. In AML models of oncogenic addiction to RAS-MEK signaling, the MEK inhibitor trametinib demonstrated antileukemic activity in vitro and in vivo. However, the efficacy of trametinib was heterogeneous in ex vivo cultures of primary RAS+ AML patient specimens. From repurposing drug screens in RAS-activated AML cells, we identified pyrvinium pamoate, an anti-helminthic agent efficiently inhibiting the growth of RAS+ primary AML cells ex vivo, preferentially in trametinib-resistant PTPN11- or KRAS-mutated samples. Metabolic and genetic complementarity between trametinib and pyrvinium pamoate translated into anti-AML synergy in vitro. Moreover, this combination inhibited the propagation of RA+ AML cells in vivo in mice, indicating a potential for future clinical development of this strategy in AML.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Combination of RSK inhibitor LJH-685 and FLT3 inhibitor FF-10101 promoted apoptosis and proliferation inhibition of AML cell lines.Cell Oncol (Dordr). 2022 Oct;45(5):1005-1018. doi: 10.1007/s13402-022-00703-7. Epub 2022 Aug 29. Cell Oncol (Dordr). 2022. PMID: 36036884
-
Activity of the oral mitogen-activated protein kinase kinase inhibitor trametinib in RAS-mutant relapsed or refractory myeloid malignancies.Cancer. 2016 Jun 15;122(12):1871-9. doi: 10.1002/cncr.29986. Epub 2016 Mar 18. Cancer. 2016. PMID: 26990290 Free PMC article. Clinical Trial.
-
[Clinical Significance of RAS Gene Mutations in Patients with Acute Myeloid Leukemia].Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2022 Oct;30(5):1391-1396. doi: 10.19746/j.cnki.issn.1009-2137.2022.05.014. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2022. PMID: 36208240 Chinese.
-
Quizartinib (AC220): a promising option for acute myeloid leukemia.Drug Des Devel Ther. 2019 Apr 8;13:1117-1125. doi: 10.2147/DDDT.S198950. eCollection 2019. Drug Des Devel Ther. 2019. PMID: 31114157 Free PMC article. Review.
-
Comprehensive Mutation Profile in Acute Myeloid Leukemia Patients with RUNX1-RUNX1T1 or CBFB-MYH11 Fusions.Turk J Haematol. 2022 Jun 1;39(2):84-93. doi: 10.4274/tjh.galenos.2022.2021.0641. Epub 2022 Apr 21. Turk J Haematol. 2022. PMID: 35445594 Free PMC article. Review.
Cited by
-
The oncogenic potential of Rab-like protein 1A (RBEL1A) GTPase: The first review of RBEL1A research with future research directions and challenges.J Cancer. 2023 Oct 2;14(17):3214-3226. doi: 10.7150/jca.84267. eCollection 2023. J Cancer. 2023. PMID: 37928422 Free PMC article. Review.
-
Activity of trametinib as maintenance therapy after allogeneic hematopoietic stem cell transplantation in patients with relapsed or refractory RAS pathway-mutated hematologic malignancies.Ann Hematol. 2025 Feb;104(2):1295-1301. doi: 10.1007/s00277-024-06046-7. Epub 2024 Oct 19. Ann Hematol. 2025. PMID: 39425789 Free PMC article. No abstract available.
-
ADAR1 is required for acute myeloid leukemia cell survival by modulating post-transcriptional Wnt signaling through impairing miRNA biogenesis.Leukemia. 2025 Mar;39(3):599-613. doi: 10.1038/s41375-024-02500-7. Epub 2024 Dec 19. Leukemia. 2025. PMID: 39702795
-
Targeting Stem Cells and Dysplastic Features With Dual MEK/ERK and STAT3 Suppression in Gastric Carcinogenesis.Gastroenterology. 2024 Jan;166(1):117-131. doi: 10.1053/j.gastro.2023.09.040. Epub 2023 Oct 4. Gastroenterology. 2024. PMID: 37802423 Free PMC article.
-
A structure-based modelling approach identifies effective drug combinations for RAS-mutant acute myeloid leukemia.bioRxiv [Preprint]. 2025 May 3:2025.04.29.651188. doi: 10.1101/2025.04.29.651188. bioRxiv. 2025. PMID: 40654850 Free PMC article. Preprint.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
