

Crucial role and mechanism of transcription-coupled DNA repair in bacteria
- PMID: 35355008
- PMCID: PMC9370829
- DOI: 10.1038/s41586-022-04530-6
Crucial role and mechanism of transcription-coupled DNA repair in bacteria
Abstract
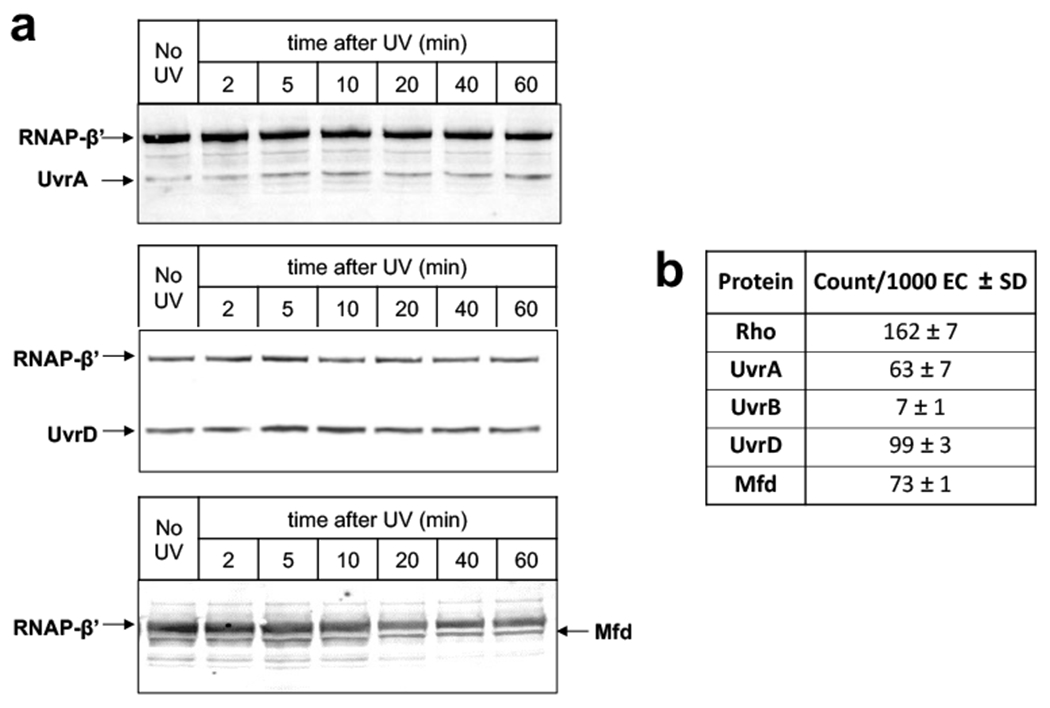
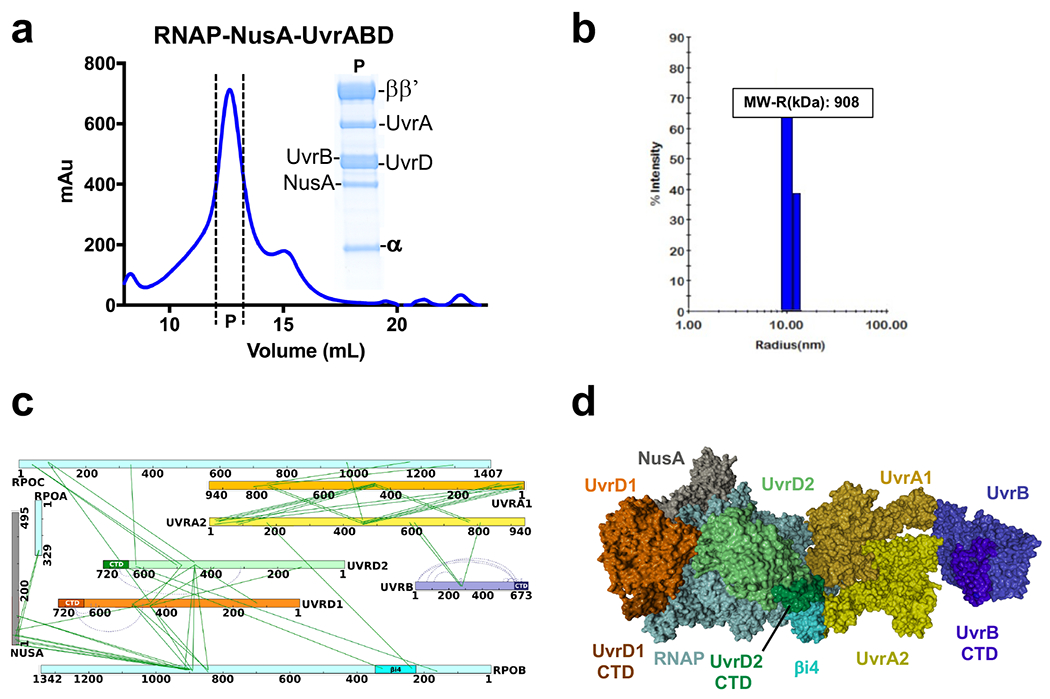
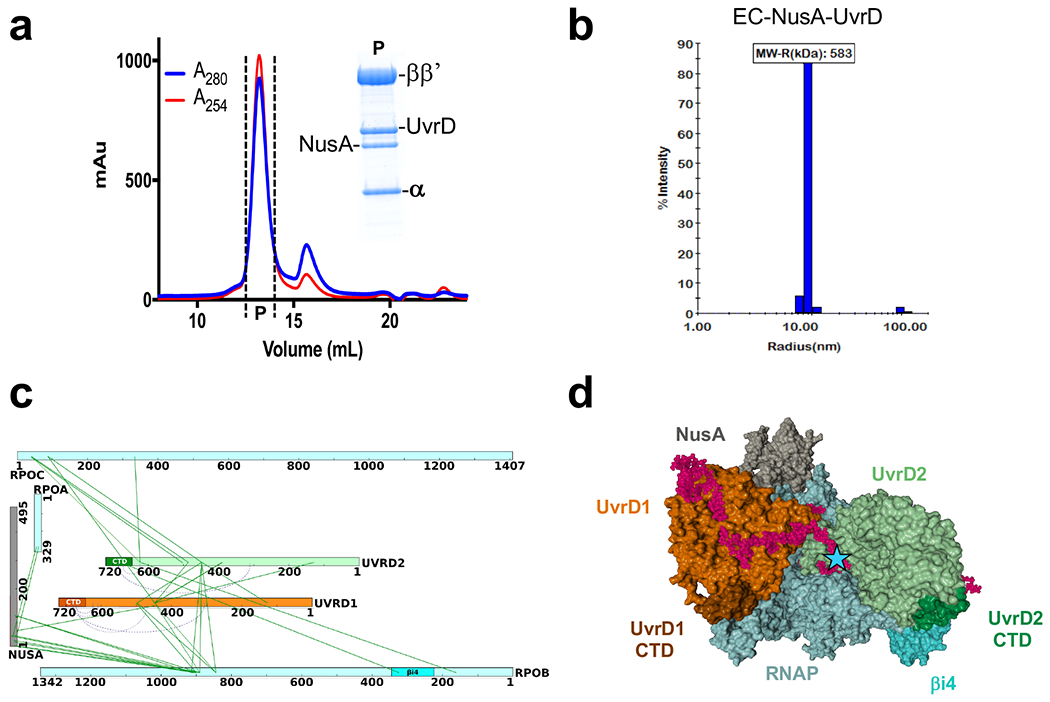
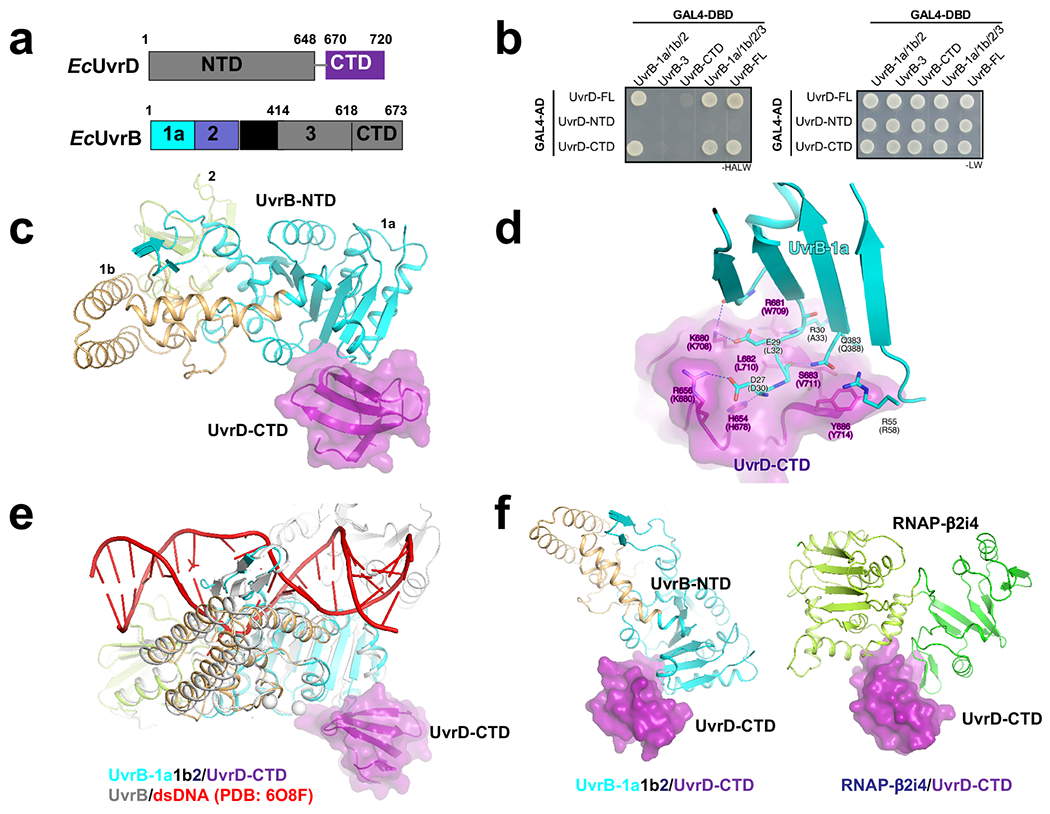
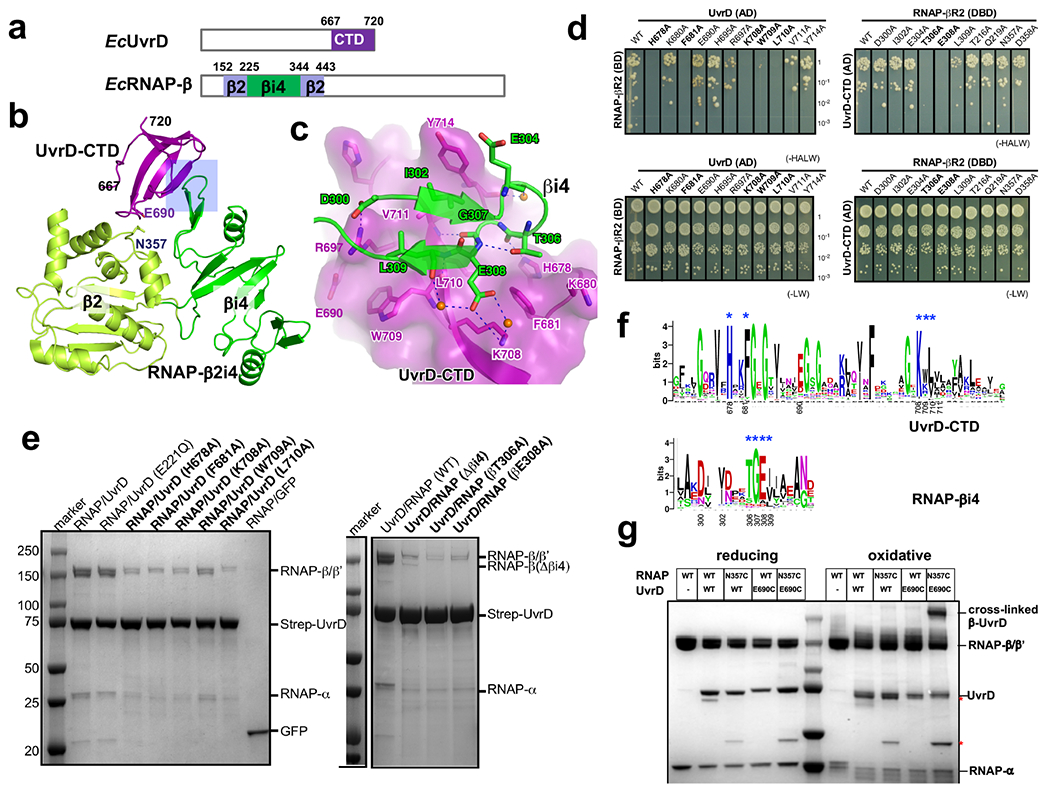
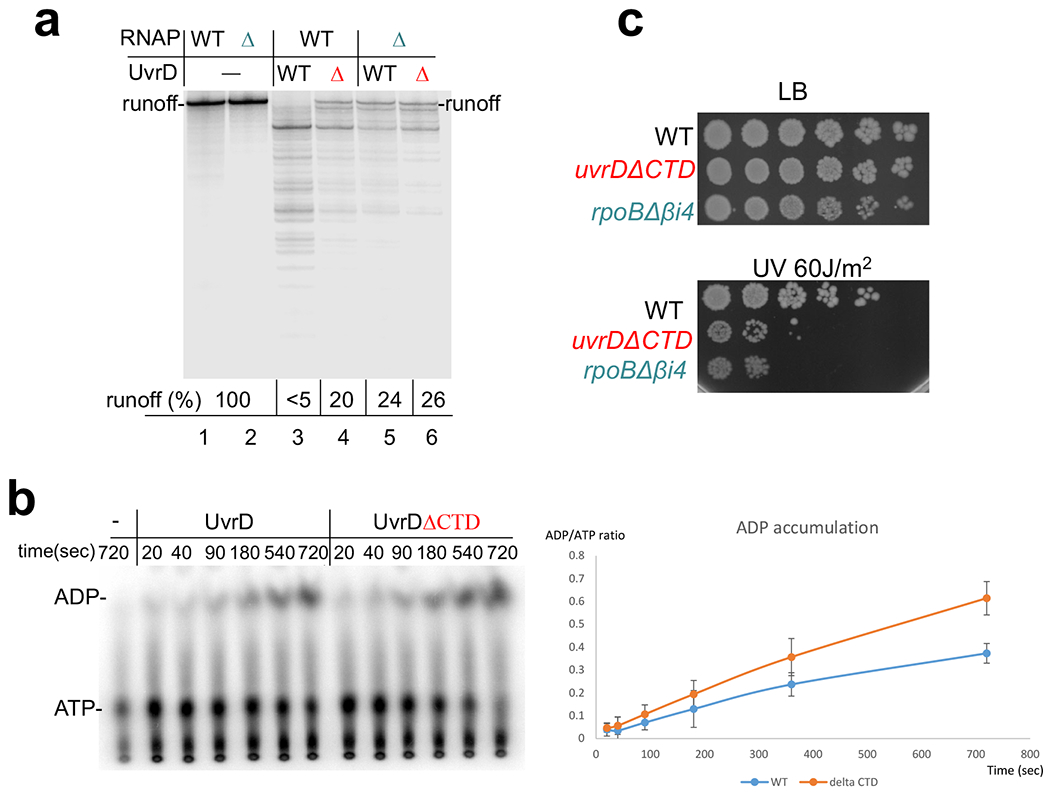
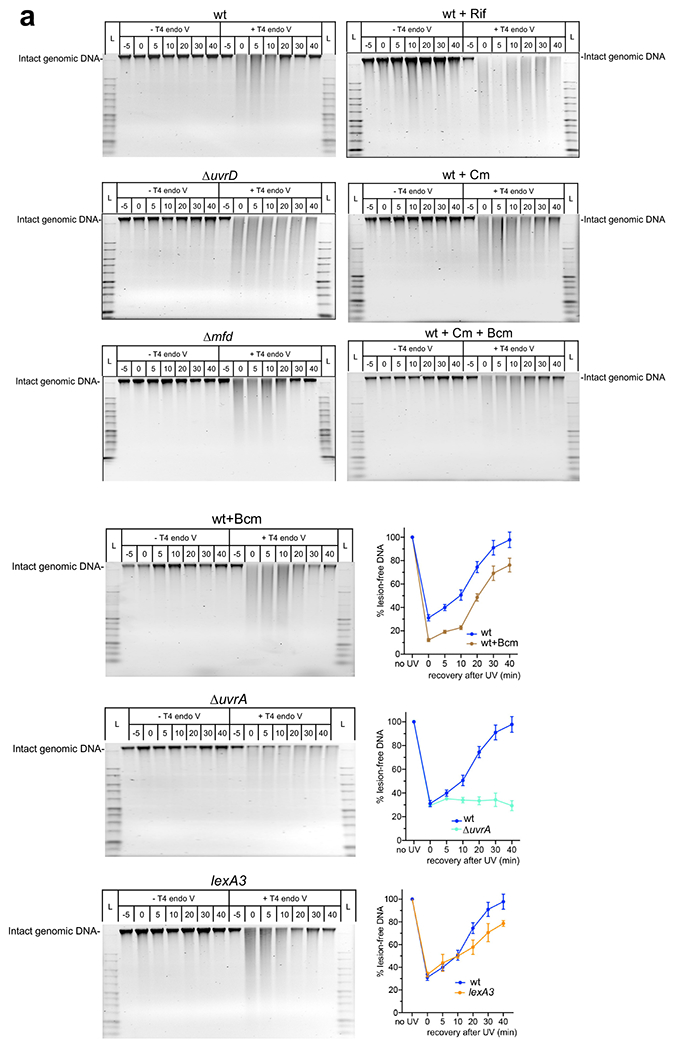
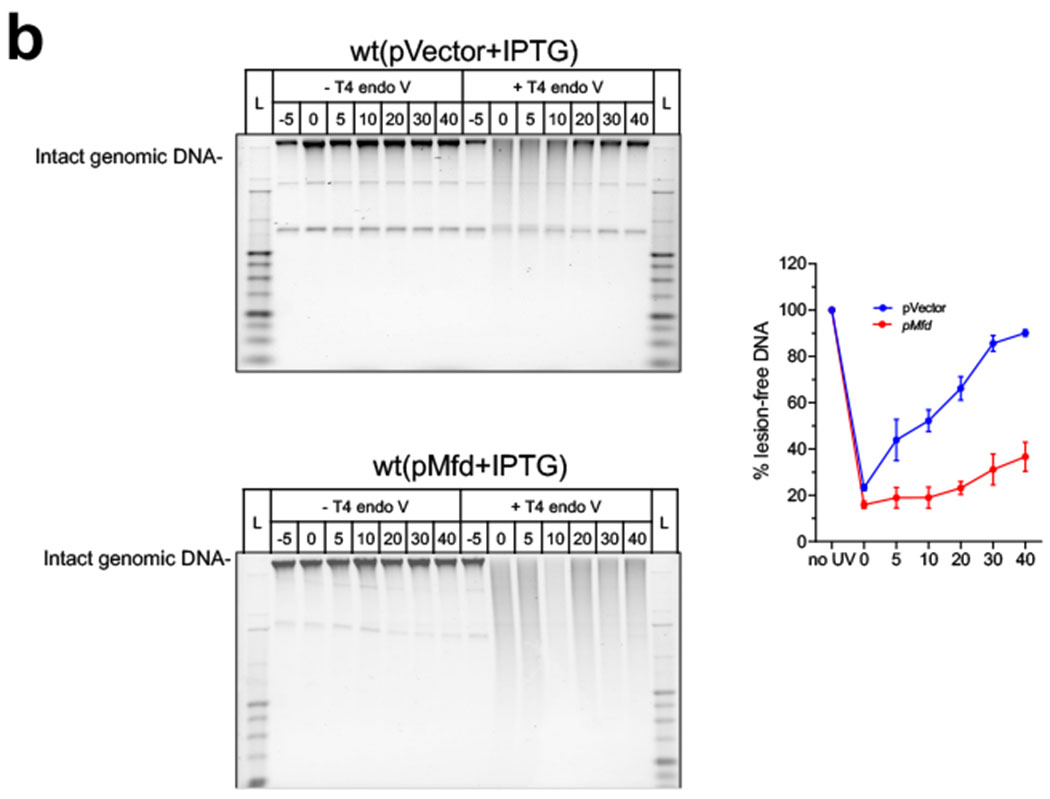
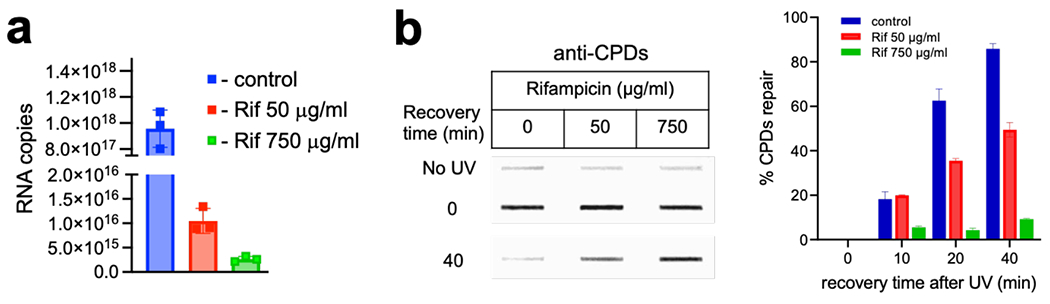
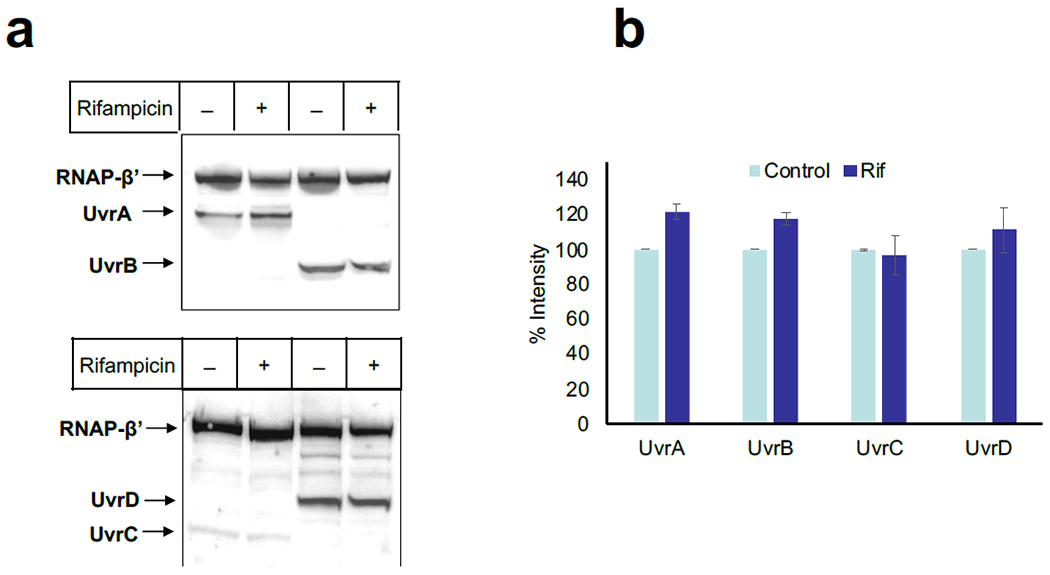
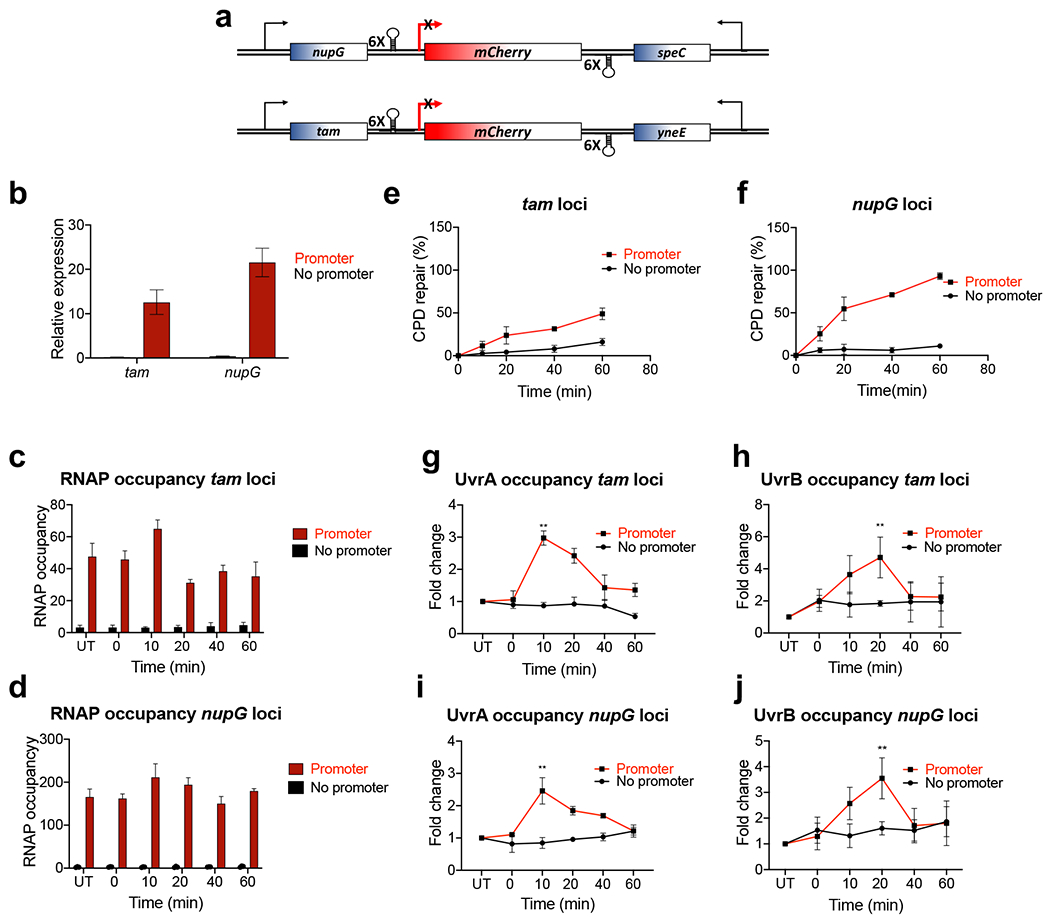
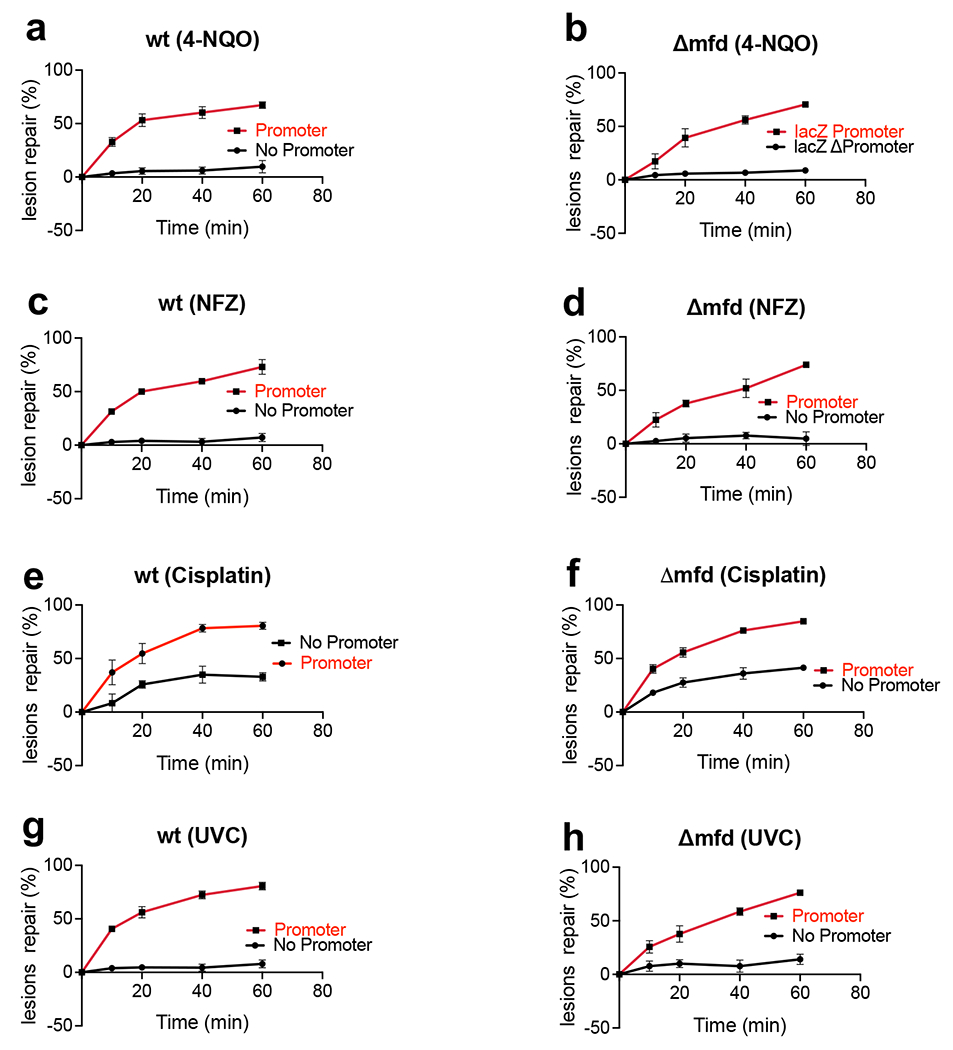
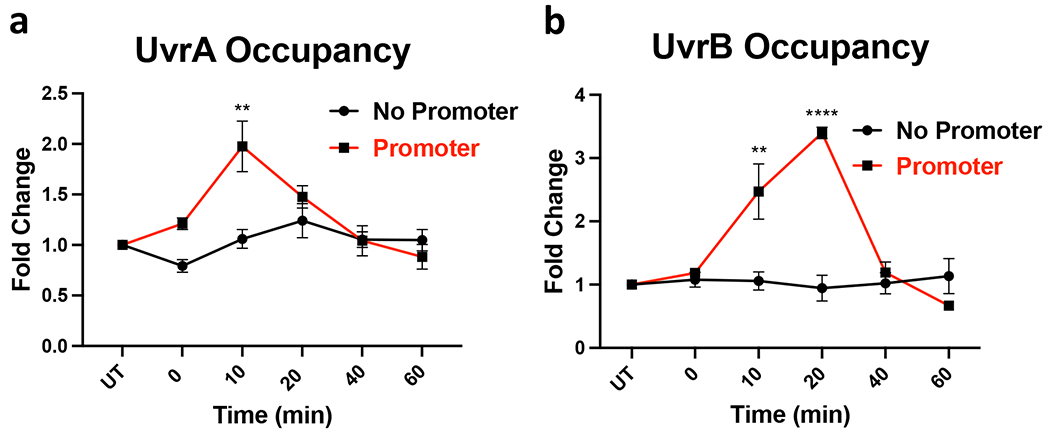
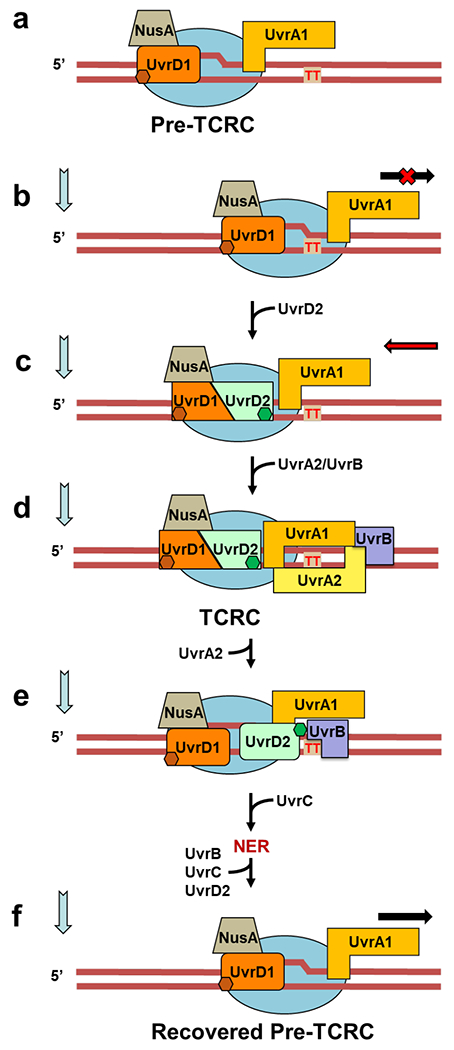
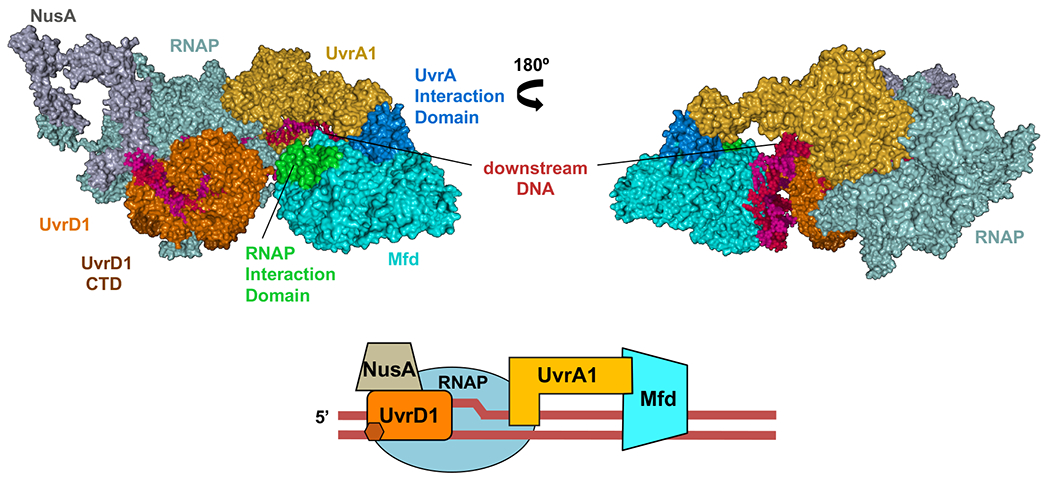
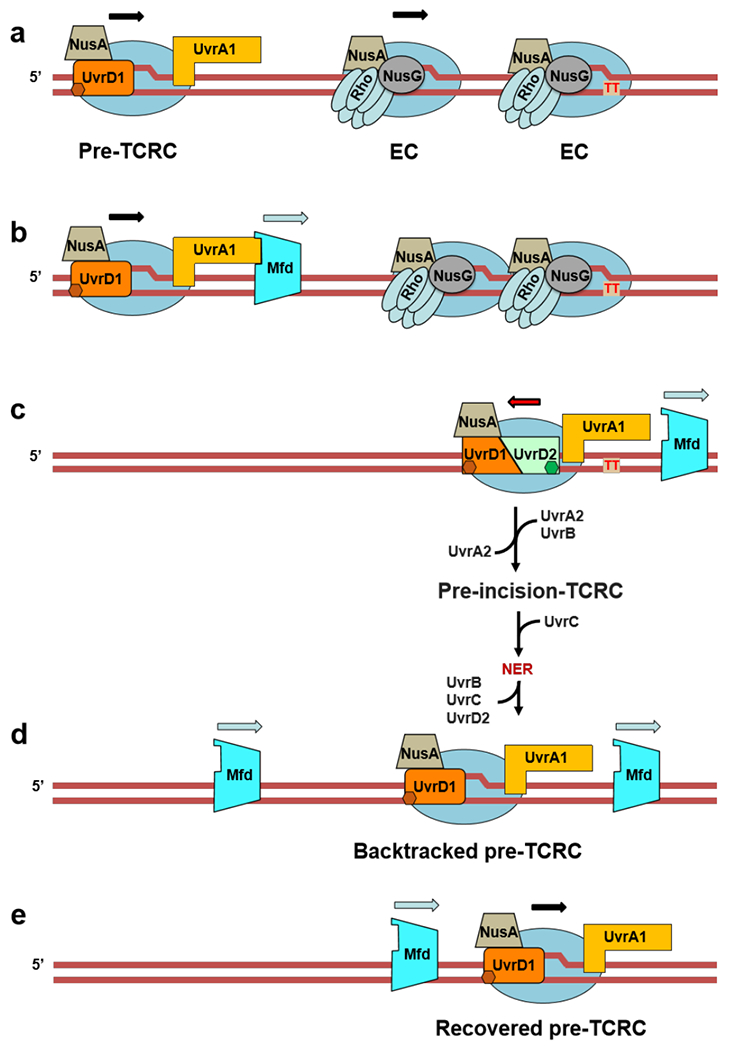
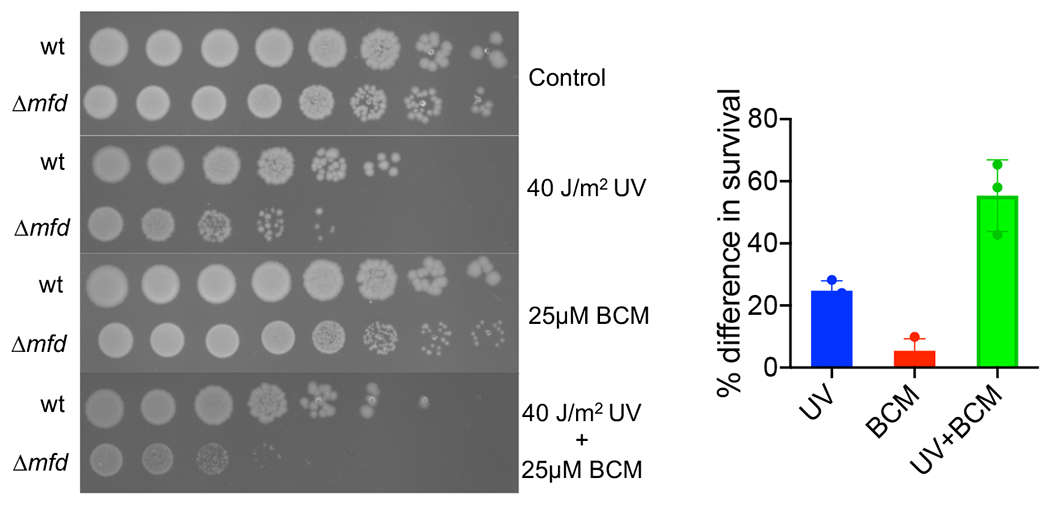
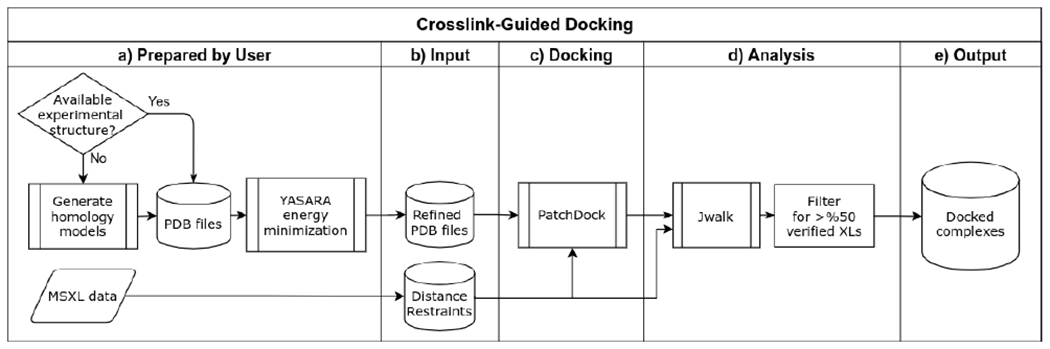
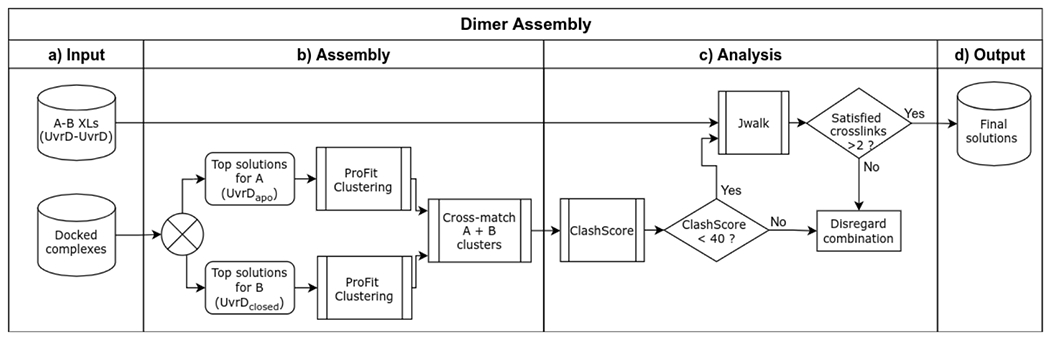
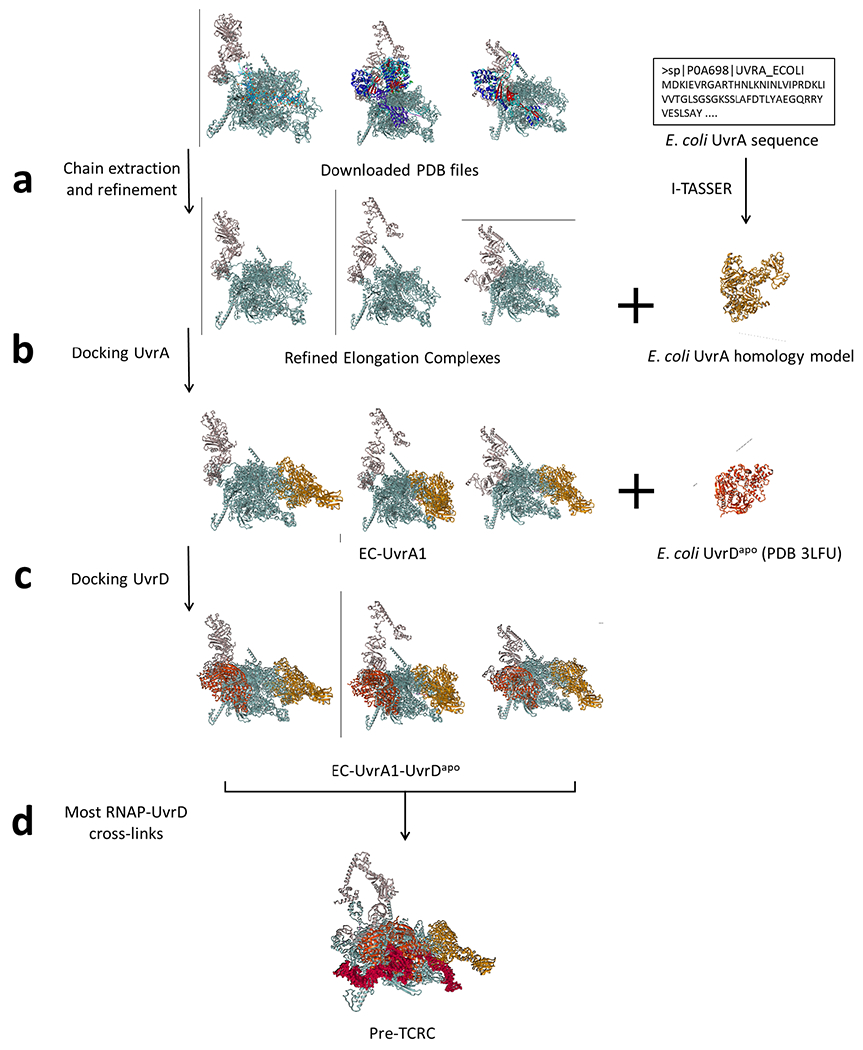
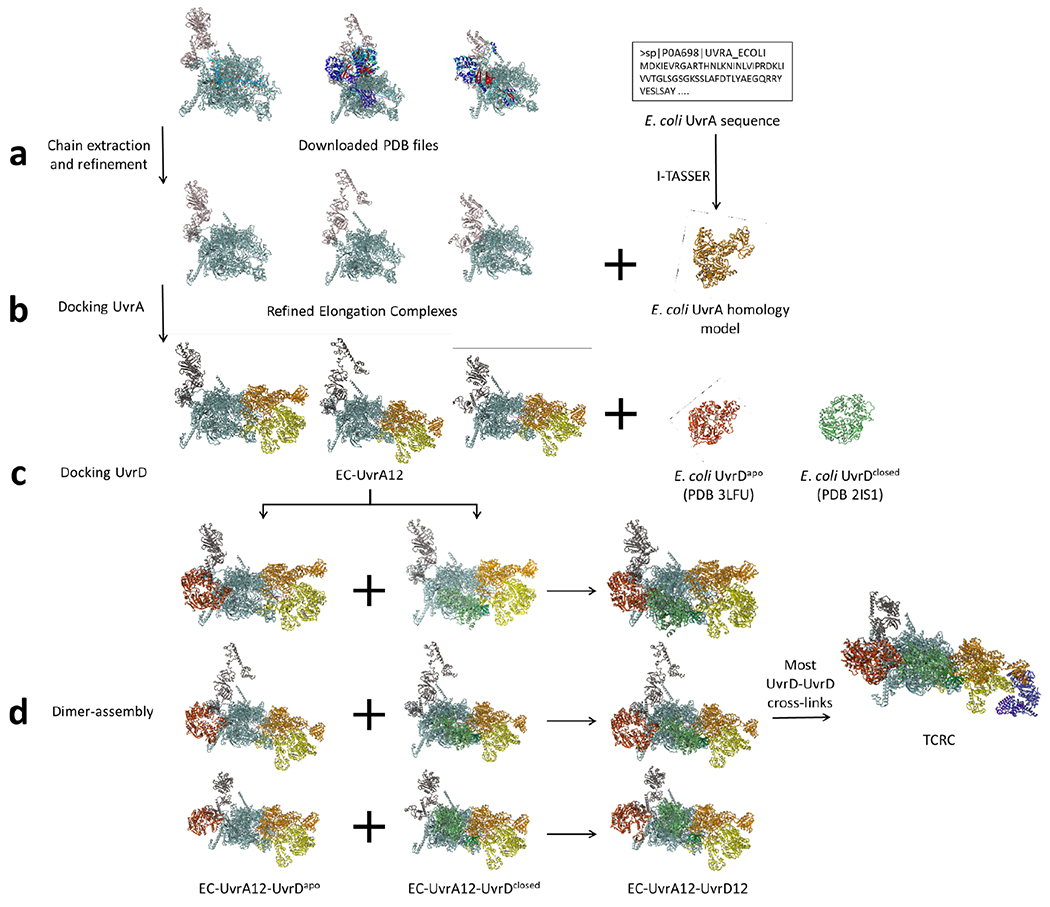
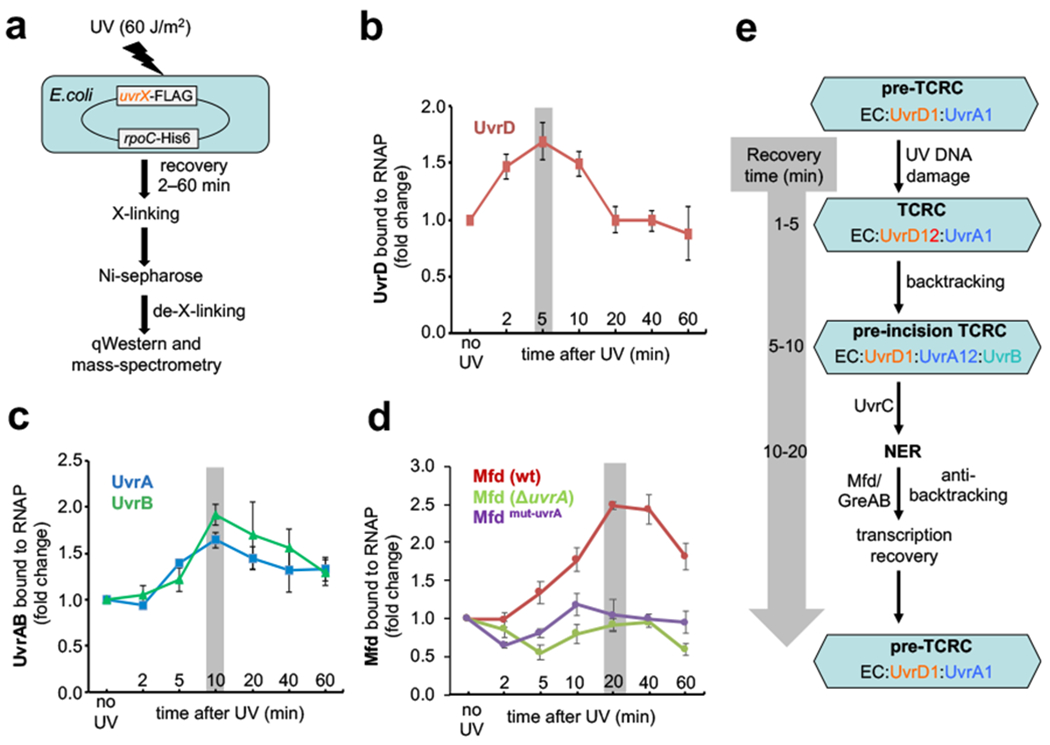
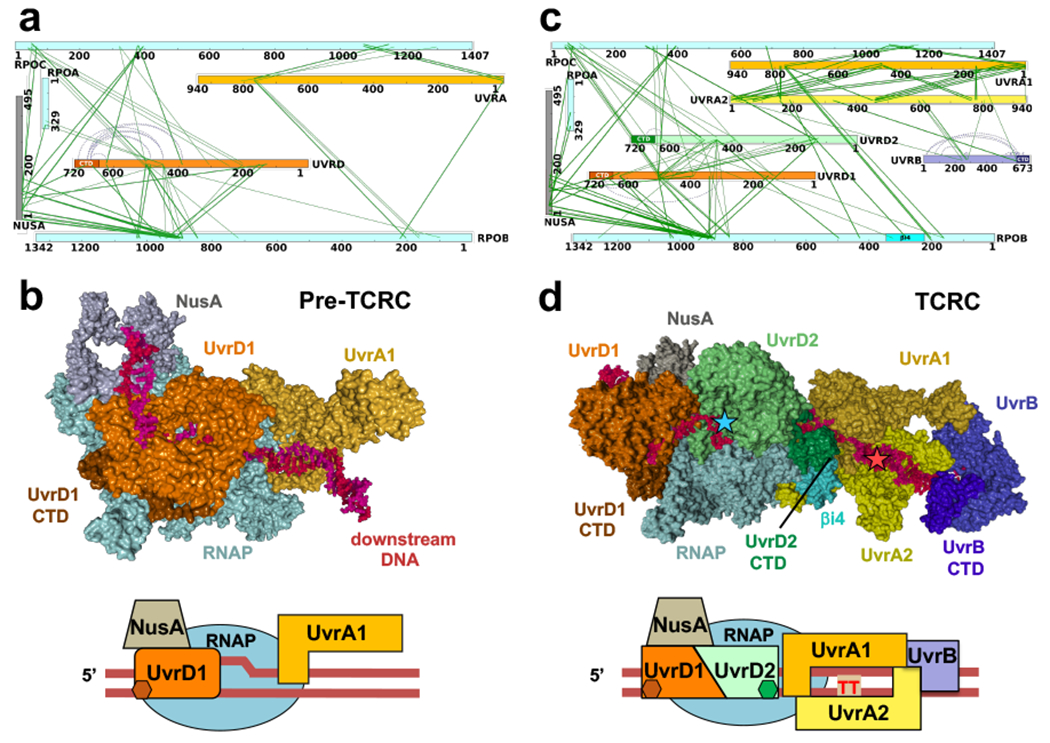
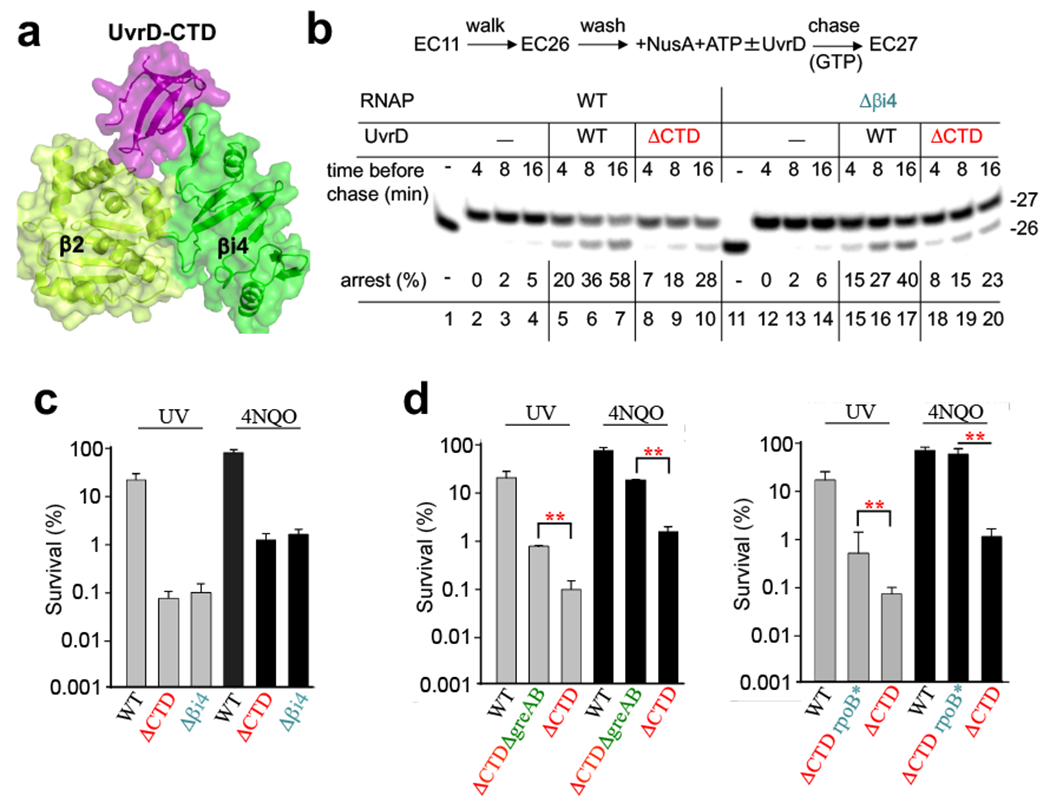
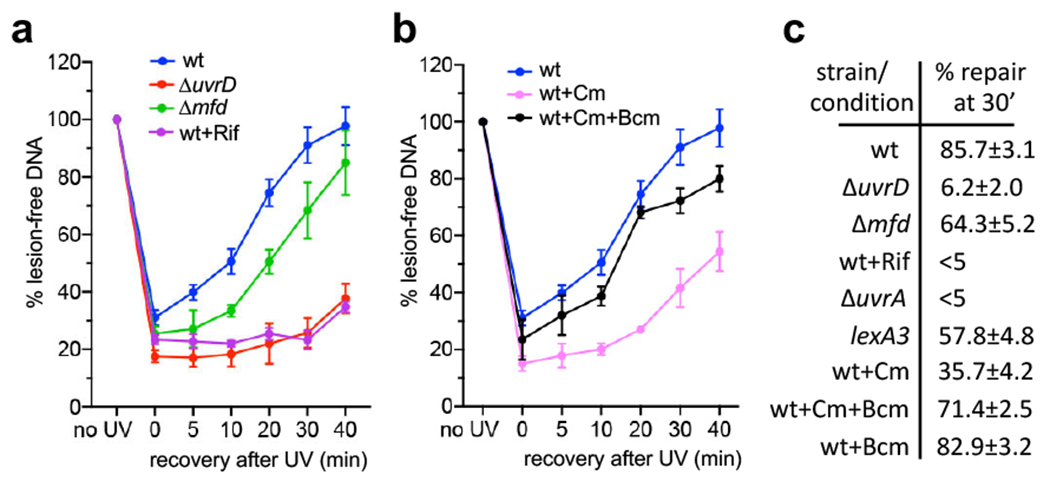
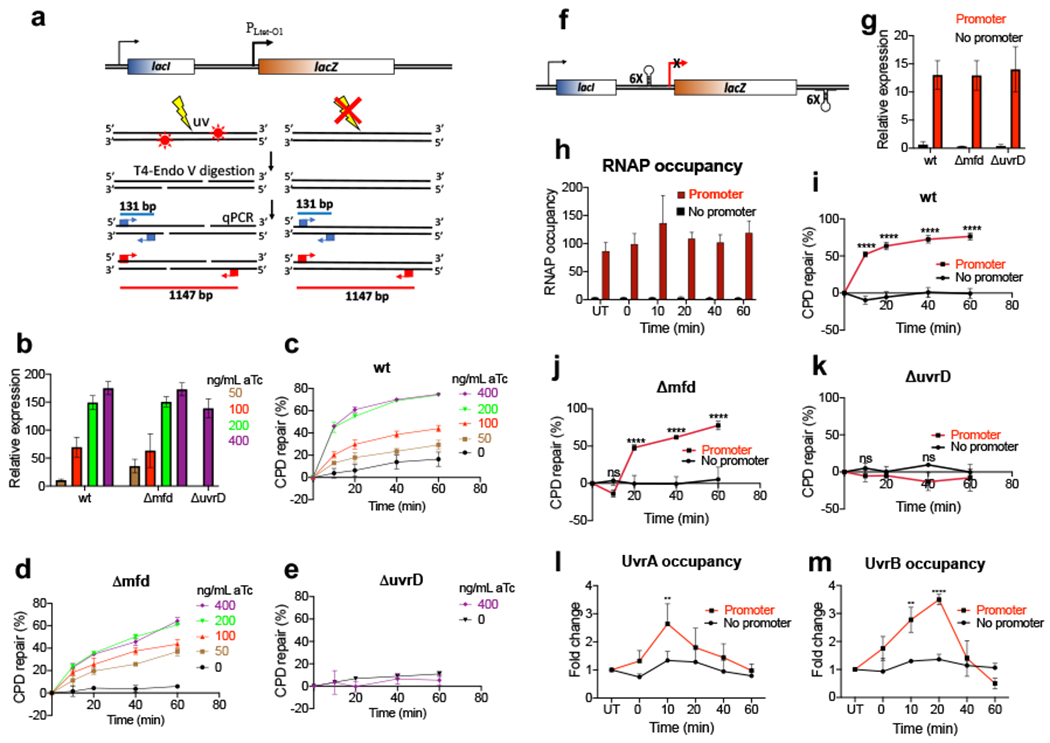
Transcription-coupled DNA repair (TCR) is presumed to be a minor sub-pathway of nucleotide excision repair (NER) in bacteria. Global genomic repair is thought to perform the bulk of repair independently of transcription. TCR is also believed to be mediated exclusively by Mfd-a DNA translocase of a marginal NER phenotype1-3. Here we combined in cellulo cross-linking mass spectrometry with structural, biochemical and genetic approaches to map the interactions within the TCR complex (TCRC) and to determine the actual sequence of events that leads to NER in vivo. We show that RNA polymerase (RNAP) serves as the primary sensor of DNA damage and acts as a platform for the recruitment of NER enzymes. UvrA and UvrD associate with RNAP continuously, forming a surveillance pre-TCRC. In response to DNA damage, pre-TCRC recruits a second UvrD monomer to form a helicase-competent UvrD dimer that promotes backtracking of the TCRC. The weakening of UvrD-RNAP interactions renders cells sensitive to genotoxic stress. TCRC then recruits a second UvrA molecule and UvrB to initiate the repair process. Contrary to the conventional view, we show that TCR accounts for the vast majority of chromosomal repair events; that is, TCR thoroughly dominates over global genomic repair. We also show that TCR is largely independent of Mfd. We propose that Mfd has an indirect role in this process: it participates in removing obstructive RNAPs in front of TCRCs and also in recovering TCRCs from backtracking after repair has been completed.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Figures

Similar articles
-
Comparing Mfd- and UvrD-dependent models of transcription coupled DNA repair in live Escherichia coli using single-molecule tracking.DNA Repair (Amst). 2024 May;137:103665. doi: 10.1016/j.dnarep.2024.103665. Epub 2024 Mar 7. DNA Repair (Amst). 2024. PMID: 38513450
-
Regulation and rate enhancement during transcription-coupled DNA repair.Mol Cell. 2010 Dec 10;40(5):714-24. doi: 10.1016/j.molcel.2010.11.012. Mol Cell. 2010. PMID: 21145481 Free PMC article.
-
Structural basis for transcription complex disruption by the Mfd translocase.Elife. 2021 Jan 22;10:e62117. doi: 10.7554/eLife.62117. Elife. 2021. PMID: 33480355 Free PMC article.
-
Transcription-coupled global genomic repair in E. coli.Trends Biochem Sci. 2023 Oct;48(10):873-882. doi: 10.1016/j.tibs.2023.07.007. Epub 2023 Aug 7. Trends Biochem Sci. 2023. PMID: 37558547 Review.
-
Rethinking transcription coupled DNA repair.Curr Opin Microbiol. 2015 Apr;24:15-20. doi: 10.1016/j.mib.2014.12.005. Epub 2015 Jan 14. Curr Opin Microbiol. 2015. PMID: 25596348 Free PMC article. Review.
Cited by
-
New advances in cross-linking mass spectrometry toward structural systems biology.Curr Opin Chem Biol. 2023 Oct;76:102357. doi: 10.1016/j.cbpa.2023.102357. Epub 2023 Jul 3. Curr Opin Chem Biol. 2023. PMID: 37406423 Free PMC article. Review.
-
Pervasive Transcription-coupled DNA repair in E. coli.Nat Commun. 2022 Mar 30;13(1):1702. doi: 10.1038/s41467-022-28871-y. Nat Commun. 2022. PMID: 35354807 Free PMC article.
-
Identification of a novel DNA repair inhibitor using an in silico driven approach shows effective combinatorial activity with genotoxic agents against multidrug-resistant Escherichia coli.Protein Sci. 2024 Apr;33(4):e4948. doi: 10.1002/pro.4948. Protein Sci. 2024. PMID: 38501485 Free PMC article.
-
ASH1L-MRG15 methyltransferase deposits H3K4me3 and FACT for damage verification in nucleotide excision repair.Nat Commun. 2023 Jul 1;14(1):3892. doi: 10.1038/s41467-023-39635-7. Nat Commun. 2023. PMID: 37393406 Free PMC article.
-
Prophage terminase with tRNase activity sensitizes Salmonella enterica to oxidative stress.Science. 2024 Apr 5;384(6691):100-105. doi: 10.1126/science.adl3222. Epub 2024 Apr 4. Science. 2024. PMID: 38574144 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
