

Chromosomal instability drives convergent and divergent evolution toward advantageous inherited traits in mammalian CHO bioproduction lineages
- PMID: 35355517
- PMCID: PMC8958363
- DOI: 10.1016/j.isci.2022.104074
Chromosomal instability drives convergent and divergent evolution toward advantageous inherited traits in mammalian CHO bioproduction lineages
Abstract
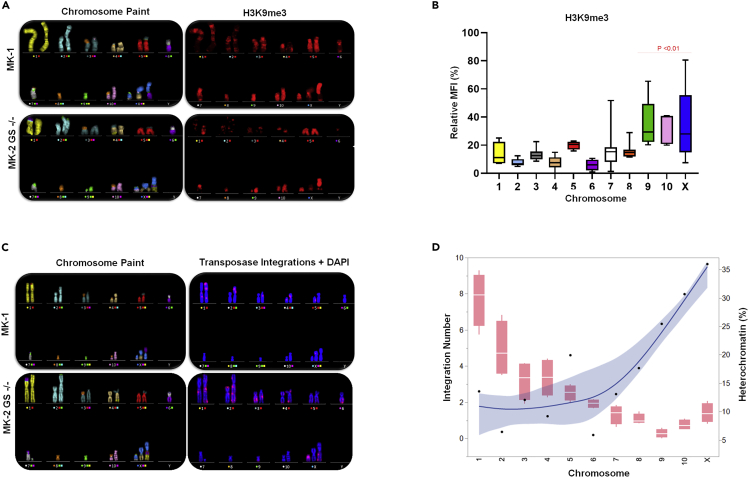
Genetic instability of Chinese hamster ovary (CHO) cells is implicated in production inconsistency through poorly defined mechanisms. Using a multi-omics approach, we analyzed the variations of CHO lineages derived from CHO-K1 cells. We identify an equilibrium between random genetic variation of the CHO genome and heritable traits driven by culture conditions, selection criteria, and genetic linkage. These inherited changes are associated with the selection pressures related to serum removal, suspension culture transition, protein expression, and secretion. We observed that a haploid reduction of a Chromosome 2 region after serum-free, suspension adaptation, was consistently inherited, suggesting common adaptation mechanisms. Genetic variations also included ∼200 insertions/deletions, ∼1000 single-nucleotide polymorphisms, and ∼300-2000 copy number variations, which were exacerbated after gene editing. In addition, heterochromatic chromosomes were preferentially lost as cells continuously evolved. Together, these observations demonstrate a highly plastic signature for adapted CHO cells and paves the way towards future host cell engineering.
Keywords: Biotechnology; Evolutionary mechanisms; Genetics; Omics.
© 2022 The Authors.
Conflict of interest statement
S. Huhn, A. Kumar, B. Jiang, R. Liu, H. Lin, G. Nyberg, and Z. Du are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA and potentially own stock and/or hold stock options in Merck & Co., Inc., Kenilworth, NJ, USA. Merck & Co., Inc provided support in the form of salaries for all authors but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the “author contributions” section.
Figures







References
-
- Borth N., Hu W.S. Enhancing CHO by systems biotechnology. Biotechnol. J. 2018;13:e1800077. - PubMed
-
- Brinkrolf K., Rupp O., Laux H., Kollin F., Ernst W., Linke B., Kofler R., Romand S., Hesse F., Budach W.E., et al. Chinese hamster genome sequenced from sorted chromosomes. Nat. Biotechnol. 2013;31:694–695. - PubMed
LinkOut - more resources
Full Text Sources
