

Non-Alcoholic Fatty Liver Disease in HIV/HBV Patients - a Metabolic Imbalance Aggravated by Antiretroviral Therapy and Perpetuated by the Hepatokine/Adipokine Axis Breakdown
- PMID: 35355551
- PMCID: PMC8959898
- DOI: 10.3389/fendo.2022.814209
Non-Alcoholic Fatty Liver Disease in HIV/HBV Patients - a Metabolic Imbalance Aggravated by Antiretroviral Therapy and Perpetuated by the Hepatokine/Adipokine Axis Breakdown
Abstract
Non-alcoholic fatty liver disease (NAFLD) is strongly associated with the metabolic syndrome and is one of the most prevalent comorbidities in HIV and HBV infected patients. HIV plays an early and direct role in the development of metabolic syndrome by disrupting the mechanism of adipogenesis and synthesis of adipokines. Adipokines, molecules that regulate the lipid metabolism, also contribute to the progression of NAFLD either directly or via hepatic organokines (hepatokines). Most hepatokines play a direct role in lipid homeostasis and liver inflammation but their role in the evolution of NAFLD is not well defined. The role of HBV in the pathogenesis of NAFLD is controversial. HBV has been previously associated with a decreased level of triglycerides and with a protective role against the development of steatosis and metabolic syndrome. At the same time HBV displays a high fibrogenetic and oncogenetic potential. In the HIV/HBV co-infection, the metabolic changes are initiated by mitochondrial dysfunction as well as by the fatty overload of the liver, two interconnected mechanisms. The evolution of NAFLD is further perpetuated by the inflammatory response to these viral agents and by the variable toxicity of the antiretroviral therapy. The current article discusses the pathogenic changes and the contribution of the hepatokine/adipokine axis in the development of NAFLD as well as the implications of HIV and HBV infection in the breakdown of the hepatokine/adipokine axis and NAFLD progression.
Keywords: HIV; adipokines; antiretroviral treatment; hepatitis B virus; hepatokines; metabolic syndrome; non-alcoholic fatty liver disease; oxidative stress.
Copyright © 2022 Iacob and Iacob.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
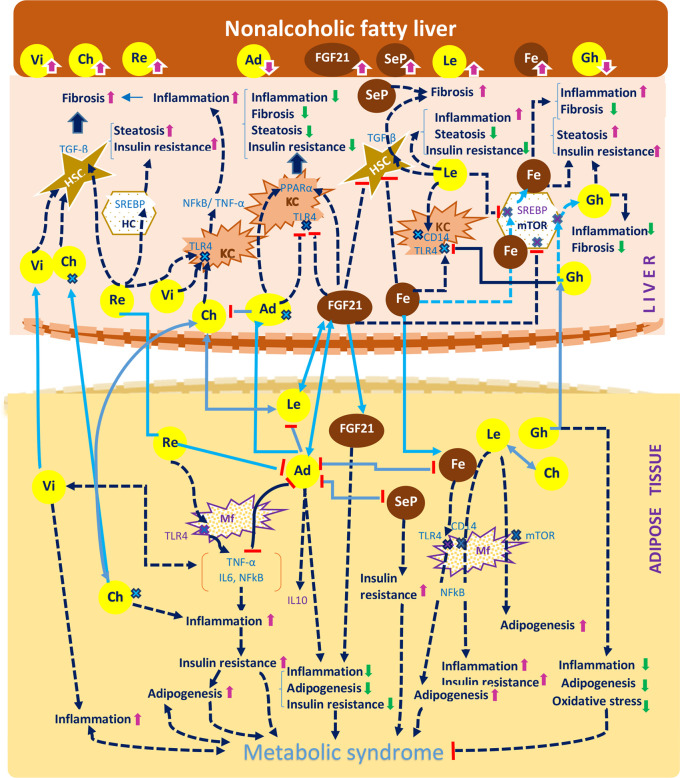
 receptor.
receptor.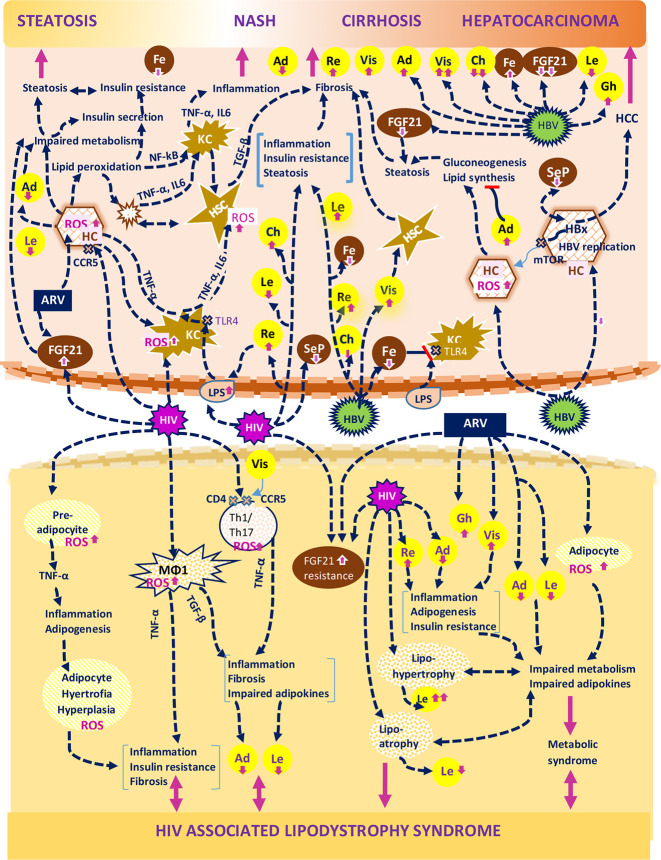
 receptor;
receptor;  apoptotic cells; TLR4, Toll-like receptor 4.
apoptotic cells; TLR4, Toll-like receptor 4.Similar articles
-
Proteomic analysis reveals exercise training induced remodelling of hepatokine secretion and uncovers syndecan-4 as a regulator of hepatic lipid metabolism.Mol Metab. 2022 Jun;60:101491. doi: 10.1016/j.molmet.2022.101491. Epub 2022 Apr 2. Mol Metab. 2022. PMID: 35381388 Free PMC article.
-
Adipokine/hepatokines profiling of fatty liver in adolescents and young adults: cross-sectional and prospective analyses of the BCAMS study.Hepatol Int. 2025 Feb;19(1):143-155. doi: 10.1007/s12072-024-10736-9. Epub 2024 Oct 14. Hepatol Int. 2025. PMID: 39400684
-
Non-alcoholic hepatic steatosis attenuates hepatitis B virus replication in an HBV-immunocompetent mouse model.Hepatol Int. 2018 Sep;12(5):438-446. doi: 10.1007/s12072-018-9877-7. Epub 2018 Jul 5. Hepatol Int. 2018. PMID: 29974410
-
Non-Alcoholic Steatohepatitis (NASH) and Organokines: What Is Now and What Will Be in the Future.Int J Mol Sci. 2022 Jan 2;23(1):498. doi: 10.3390/ijms23010498. Int J Mol Sci. 2022. PMID: 35008925 Free PMC article. Review.
-
The role of hepatokines in NAFLD.Cell Metab. 2023 Feb 7;35(2):236-252. doi: 10.1016/j.cmet.2023.01.006. Cell Metab. 2023. PMID: 36754018 Free PMC article. Review.
Cited by
-
Unravelling the Interplay: Exploring the Influence of Previous Hepatitis B Virus, Hepatitis A Virus, and Hepatitis E Virus Infections on Non-alcoholic Fatty Liver Disease.Cureus. 2023 Sep 9;15(9):e44969. doi: 10.7759/cureus.44969. eCollection 2023 Sep. Cureus. 2023. PMID: 37822444 Free PMC article. Review.
-
Impact of hepatic steatosis on mortality, hepatocellular carcinoma, end-stage liver disease and HBsAg seroclearance in chronic hepatitis B: a United States cohort study.Front Immunol. 2025 Apr 2;16:1566925. doi: 10.3389/fimmu.2025.1566925. eCollection 2025. Front Immunol. 2025. PMID: 40255390 Free PMC article.
-
Fuzi-Lizhong Decoction Alleviates Nonalcoholic Fatty Liver Disease by Blocking TLR4/MyD88/TRAF6 Signaling.Evid Based Complement Alternat Med. 2022 Aug 26;2022:1637701. doi: 10.1155/2022/1637701. eCollection 2022. Evid Based Complement Alternat Med. 2022. PMID: 36065267 Free PMC article.
-
The protective roles of augmenter of liver regeneration in hepatocytes in the non-alcoholic fatty liver disease.Front Pharmacol. 2022 Oct 11;13:928606. doi: 10.3389/fphar.2022.928606. eCollection 2022. Front Pharmacol. 2022. PMID: 36304168 Free PMC article. Review.
-
Risk Factors Related to the Development of Nonalcoholic Fatty Liver: A Systematic Review.Can J Gastroenterol Hepatol. 2025 Apr 15;2025:9964486. doi: 10.1155/cjgh/9964486. eCollection 2025. Can J Gastroenterol Hepatol. 2025. PMID: 40264655 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical