

Unifying the known and unknown microbial coding sequence space
- PMID: 35356891
- PMCID: PMC9132574
- DOI: 10.7554/eLife.67667
Unifying the known and unknown microbial coding sequence space
Abstract
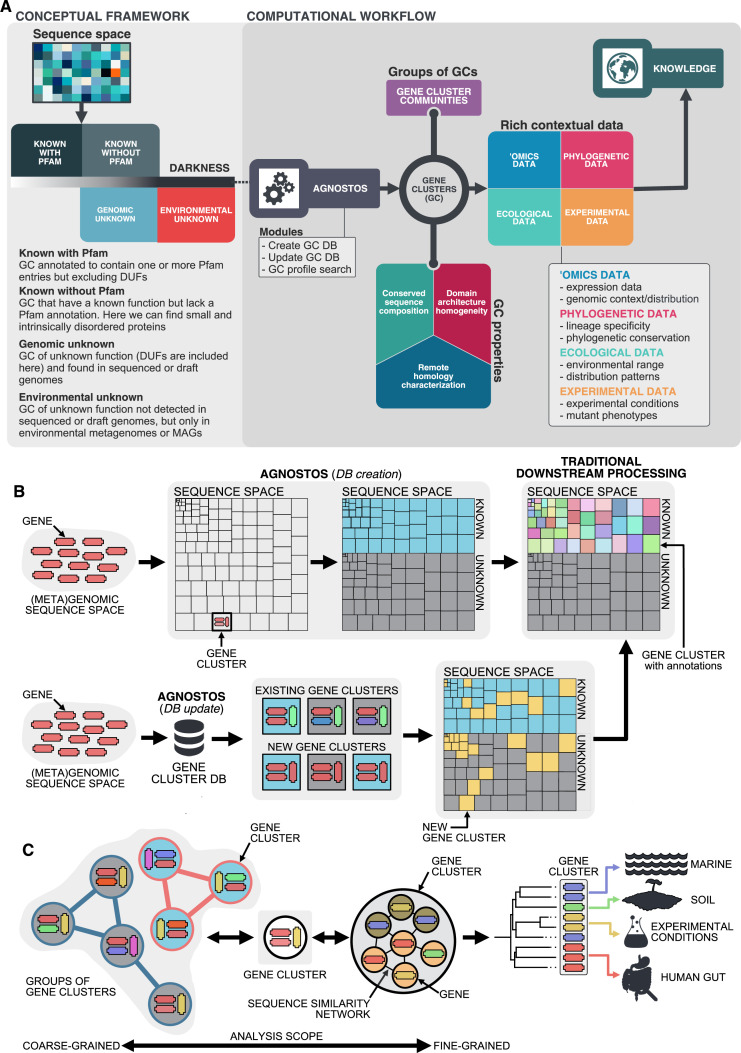
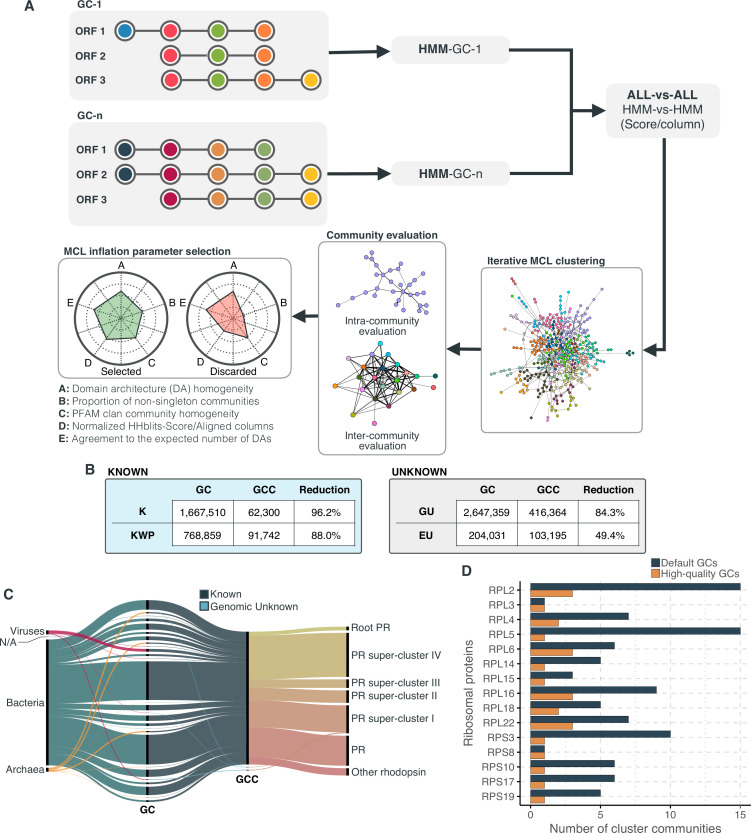
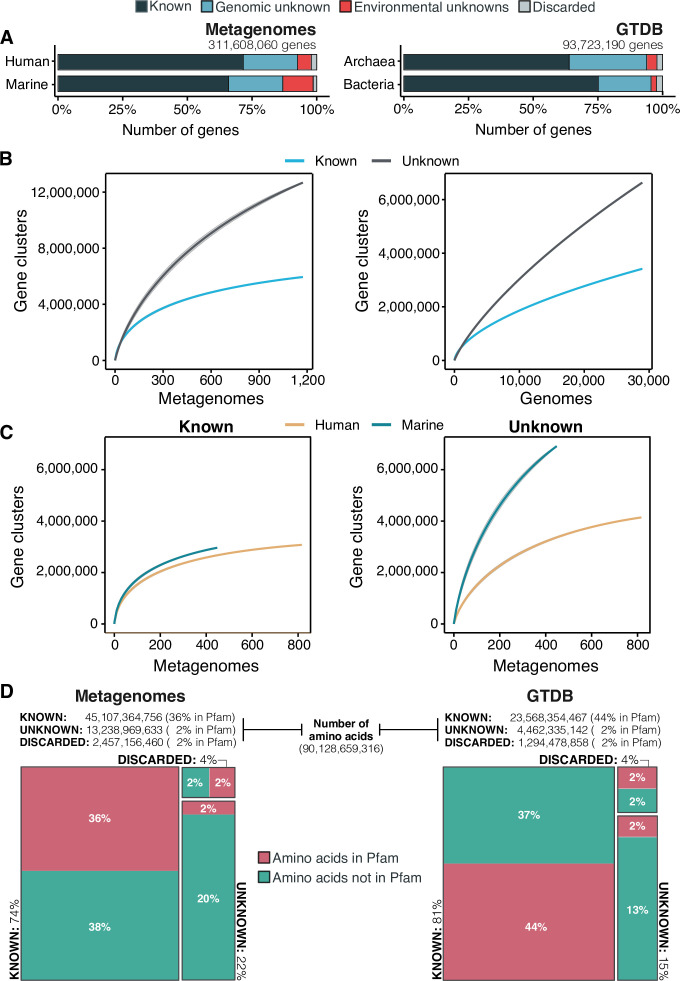
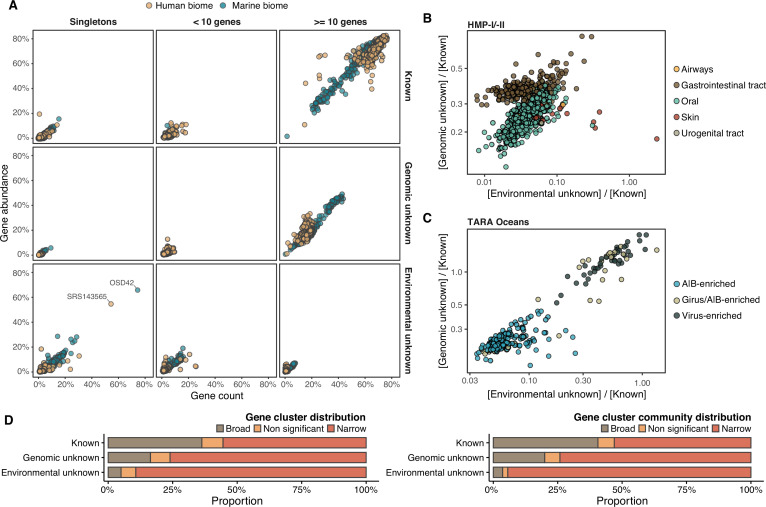
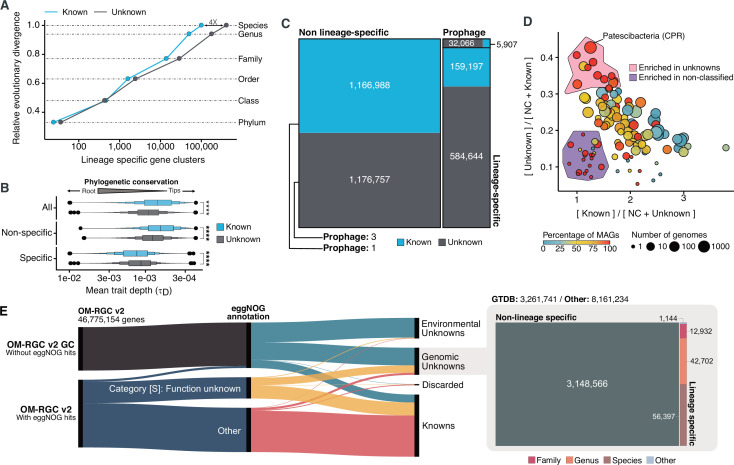
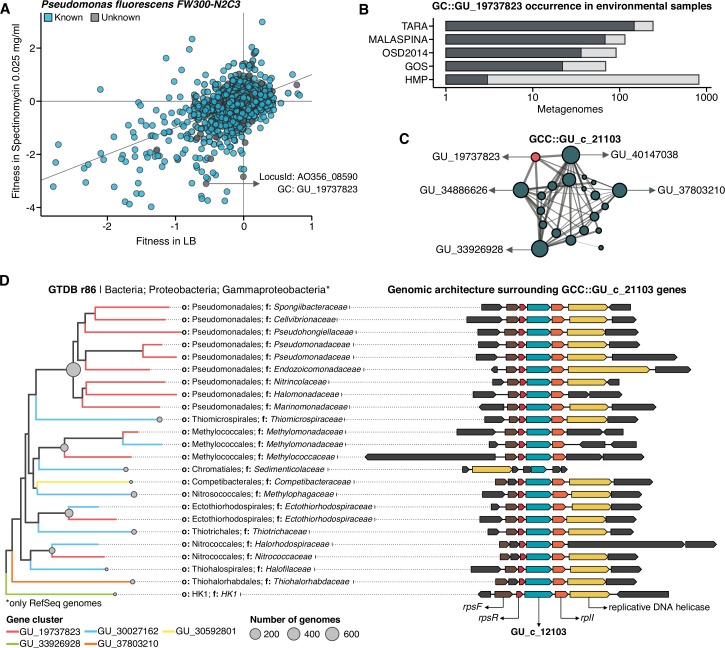
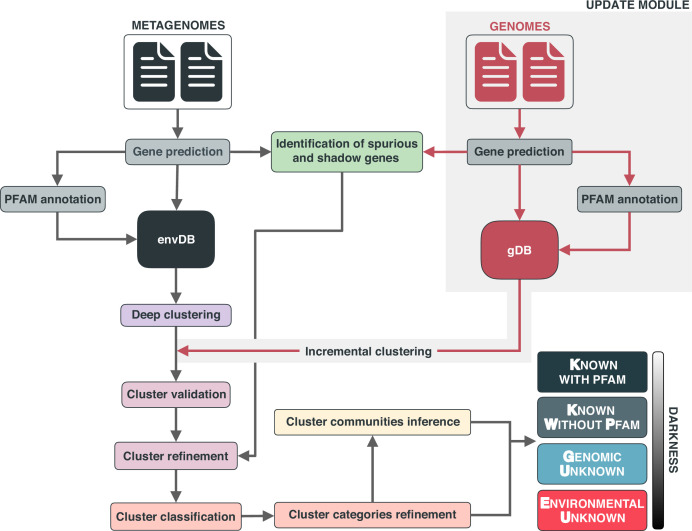
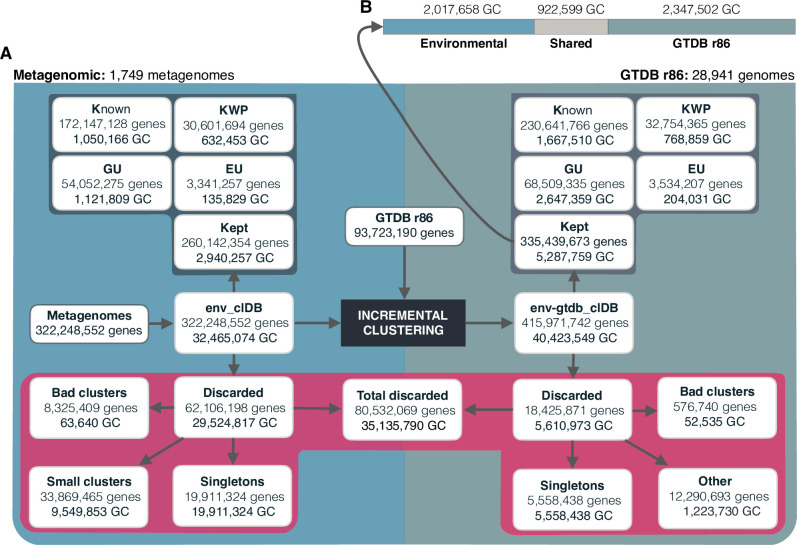
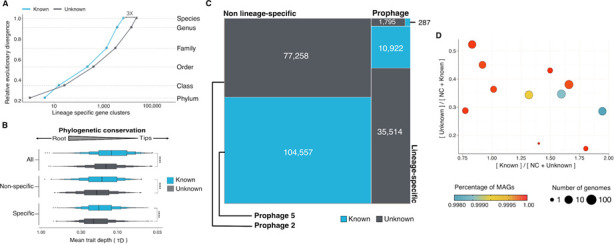
Genes of unknown function are among the biggest challenges in molecular biology, especially in microbial systems, where 40-60% of the predicted genes are unknown. Despite previous attempts, systematic approaches to include the unknown fraction into analytical workflows are still lacking. Here, we present a conceptual framework, its translation into the computational workflow AGNOSTOS and a demonstration on how we can bridge the known-unknown gap in genomes and metagenomes. By analyzing 415,971,742 genes predicted from 1749 metagenomes and 28,941 bacterial and archaeal genomes, we quantify the extent of the unknown fraction, its diversity, and its relevance across multiple organisms and environments. The unknown sequence space is exceptionally diverse, phylogenetically more conserved than the known fraction and predominantly taxonomically restricted at the species level. From the 71 M genes identified to be of unknown function, we compiled a collection of 283,874 lineage-specific genes of unknown function for Cand. Patescibacteria (also known as Candidate Phyla Radiation, CPR), which provides a significant resource to expand our understanding of their unusual biology. Finally, by identifying a target gene of unknown function for antibiotic resistance, we demonstrate how we can enable the generation of hypotheses that can be used to augment experimental data.
Keywords: bioinformatics; computational biology; functional metageomics; gene clusters; infectious disease; microbial genomics; microbiology; phylogenomics; systems biology; unknown function.
Plain language summary
It is estimated that scientists do not know what half of microbial genes actually do. When these genes are discovered in microorganisms grown in the lab or found in environmental samples, it is not possible to identify what their roles are. Many of these genes are excluded from further analyses for these reasons, meaning that the study of microbial genes tends to be limited to genes that have already been described. These limitations hinder research into microbiology, because information from newly discovered genes cannot be integrated to better understand how these organisms work. Experiments to understand what role these genes have in the microorganisms are labor-intensive, so new analytical strategies are needed. To do this, Vanni et al. developed a new framework to categorize genes with unknown roles, and a computational workflow to integrate them into traditional analyses. When this approach was applied to over 400 million microbial genes (both with known and unknown roles), it showed that the share of genes with unknown functions is only about 30 per cent, smaller than previously thought. The analysis also showed that these genes are very diverse, revealing a huge space for future research and potential applications. Combining their approach with experimental data, Vanni et al. were able to identify a gene with a previously unknown purpose that could be involved in antibiotic resistance. This system could be useful for other scientists studying microorganisms to get a more complete view of microbial systems. In future, it may also be used to analyze the genetics of other organisms, such as plants and animals.
© 2022, Vanni et al.
Conflict of interest statement
CV, MS, SA, AB, PB, EC, TD, CD, AE, RF, RK, AM, PS, KS, MS, FG, AF No competing interests declared
Figures
References
-
- Almeida A, Nayfach S, Boland M, Strozzi F, Beracochea M, Shi ZJ, Pollard KS, Sakharova E, Parks DH, Hugenholtz P, Segata N, Kyrpides NC, Finn RD. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nature Biotechnology. 2021;39:105–114. doi: 10.1038/s41587-020-0603-3. - DOI - PMC - PubMed
-
- Arnold FH. Design by Directed Evolution. Accounts of Chemical Research. 1998;31:125–131. doi: 10.1021/ar960017f. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
