

Epigenetic patterns in a complete human genome
- PMID: 35357915
- PMCID: PMC9170183
- DOI: 10.1126/science.abj5089
Epigenetic patterns in a complete human genome
Abstract
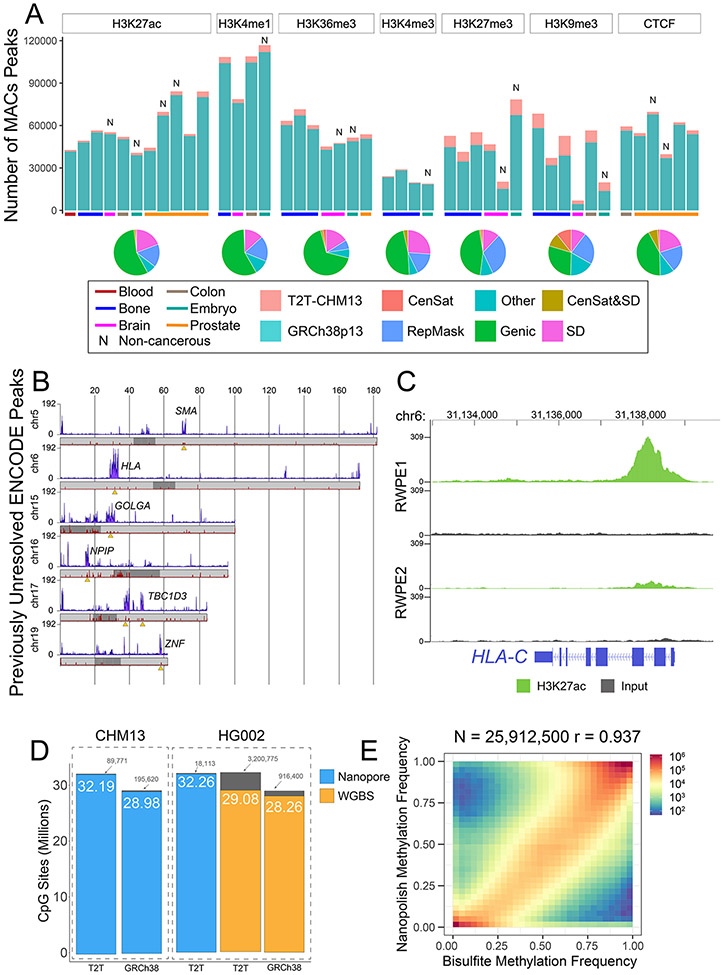
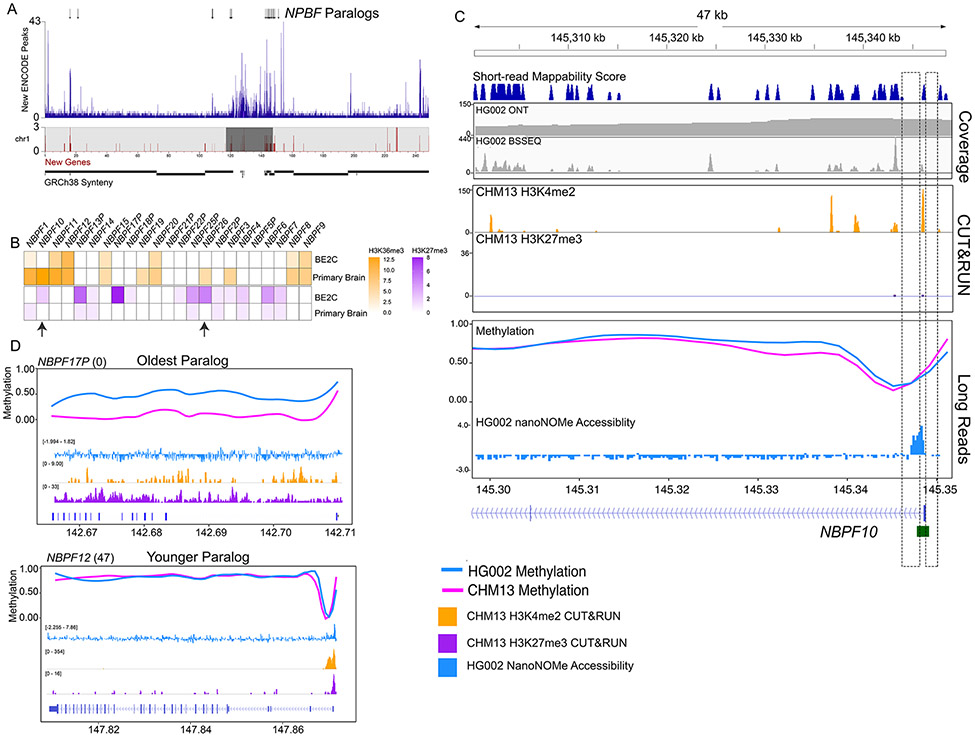
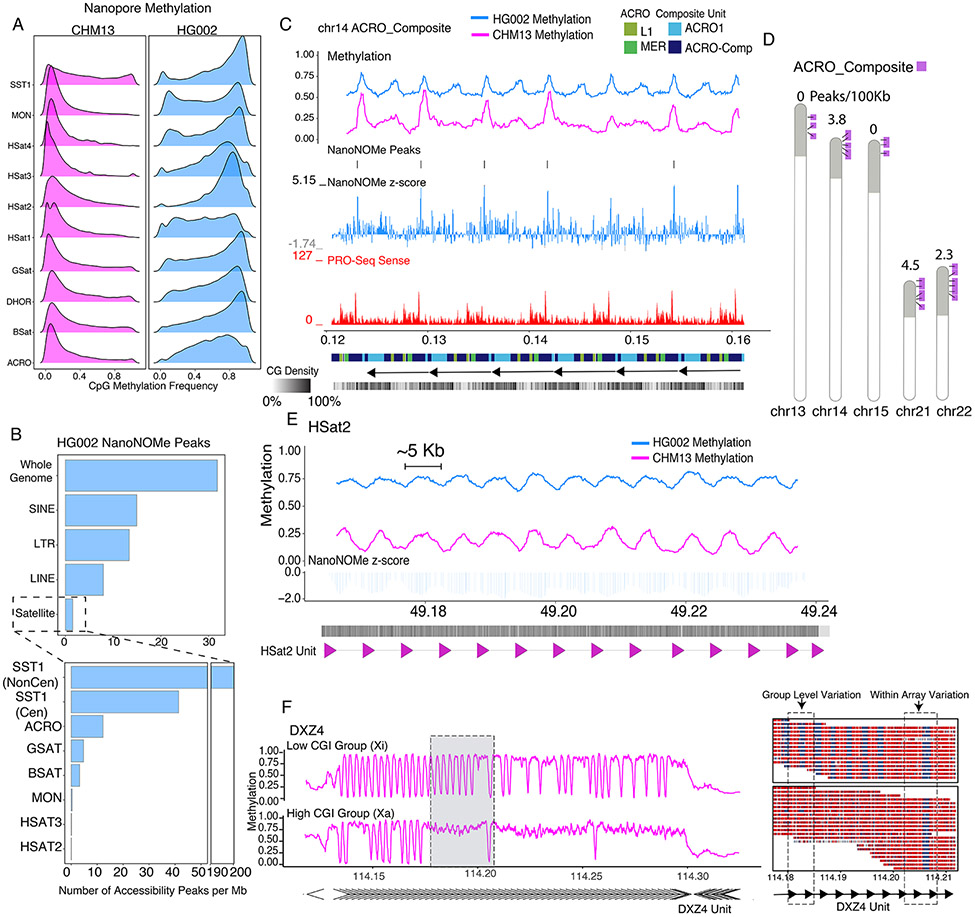
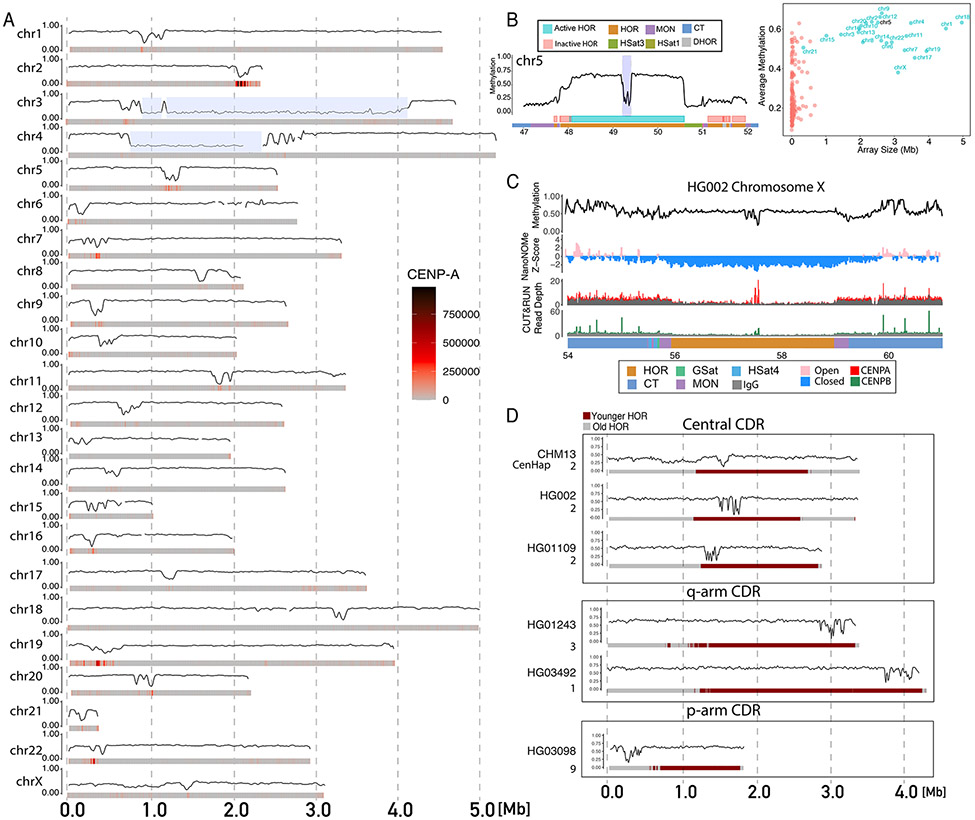
The completion of a telomere-to-telomere human reference genome, T2T-CHM13, has resolved complex regions of the genome, including repetitive and homologous regions. Here, we present a high-resolution epigenetic study of previously unresolved sequences, representing entire acrocentric chromosome short arms, gene family expansions, and a diverse collection of repeat classes. This resource precisely maps CpG methylation (32.28 million CpGs), DNA accessibility, and short-read datasets (166,058 previously unresolved chromatin immunoprecipitation sequencing peaks) to provide evidence of activity across previously unidentified or corrected genes and reveals clinically relevant paralog-specific regulation. Probing CpG methylation across human centromeres from six diverse individuals generated an estimate of variability in kinetochore localization. This analysis provides a framework with which to investigate the most elusive regions of the human genome, granting insights into epigenetic regulation.
Figures
Comment in
-
The final pieces of the human genome.Nat Rev Genet. 2022 Jun;23(6):321. doi: 10.1038/s41576-022-00494-5. Nat Rev Genet. 2022. PMID: 35488041 No abstract available.
References
-
- Nurk S et al. , The complete sequence of a human genome. bioRxiv (2021), p. 2021.05.26.445798.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
