

From telomere to telomere: The transcriptional and epigenetic state of human repeat elements
- PMID: 35357925
- PMCID: PMC9301658
- DOI: 10.1126/science.abk3112
From telomere to telomere: The transcriptional and epigenetic state of human repeat elements
Abstract
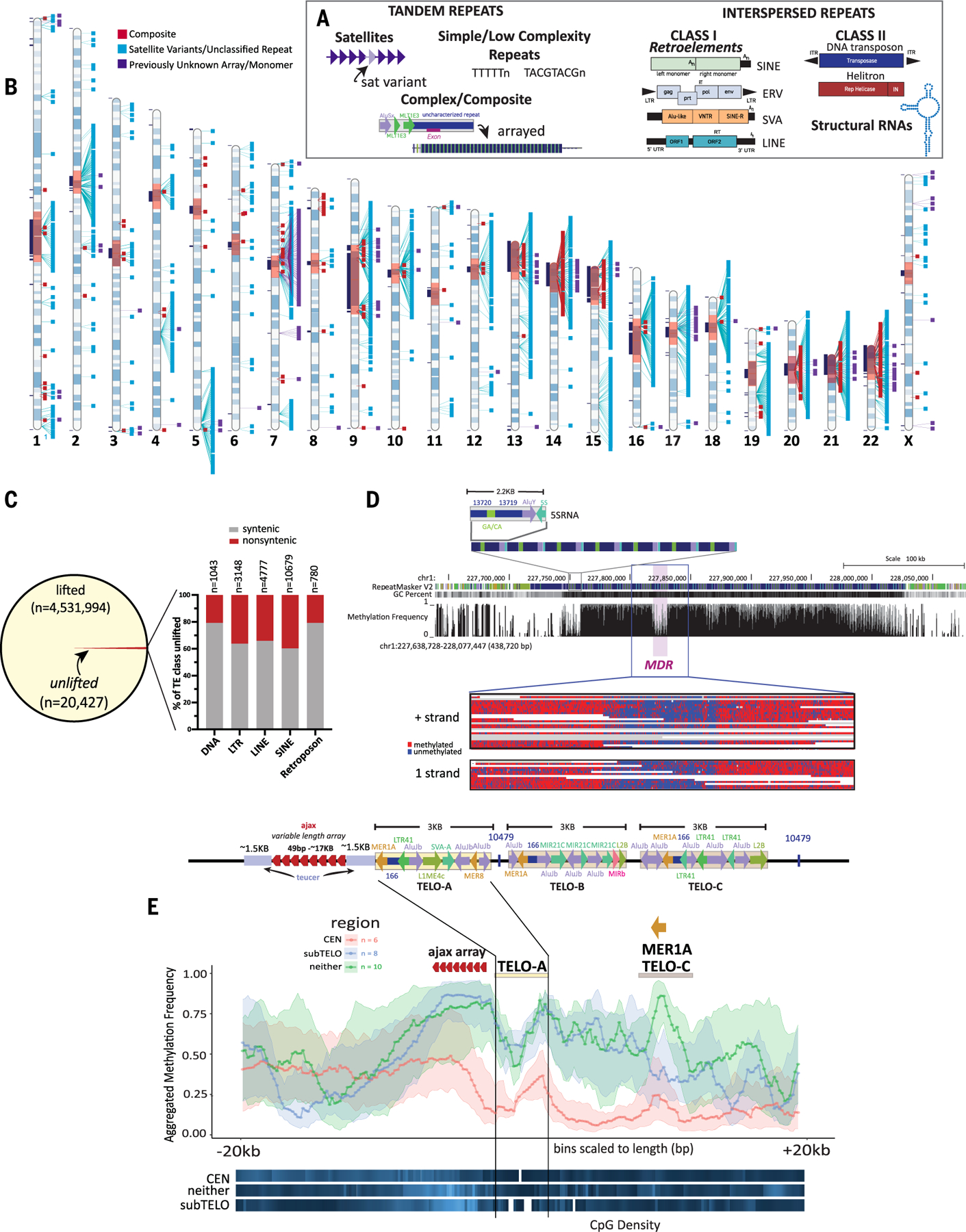
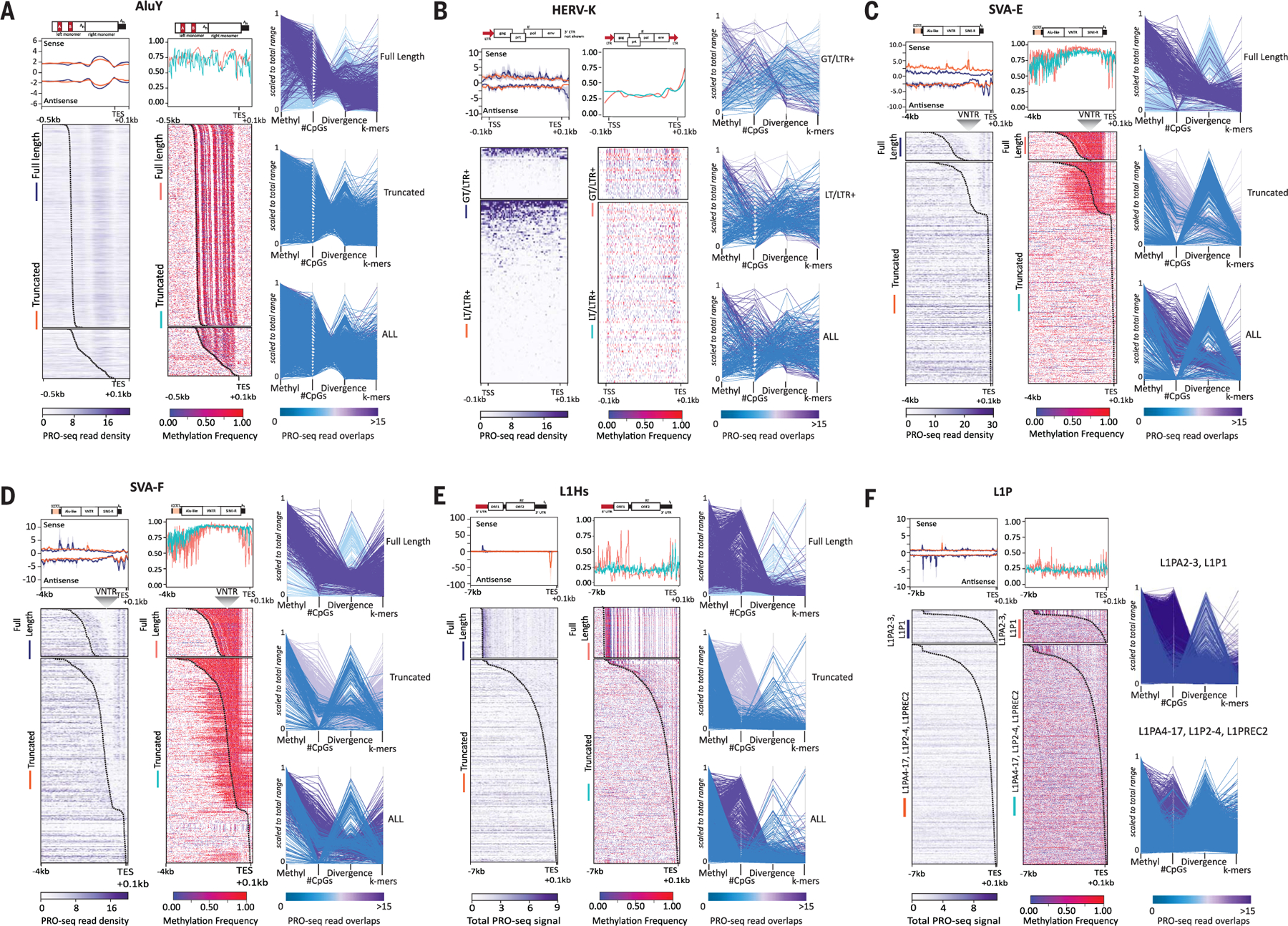
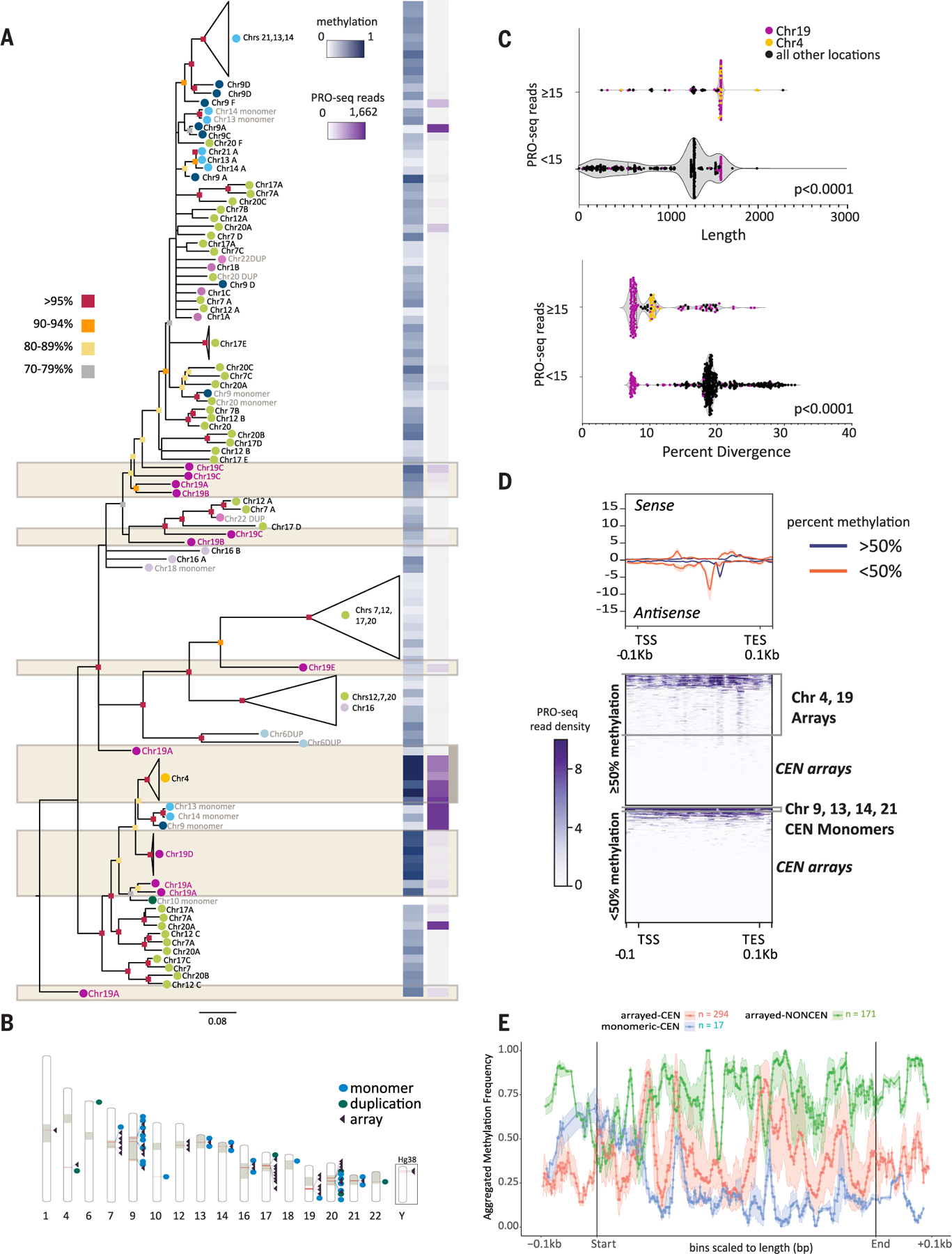
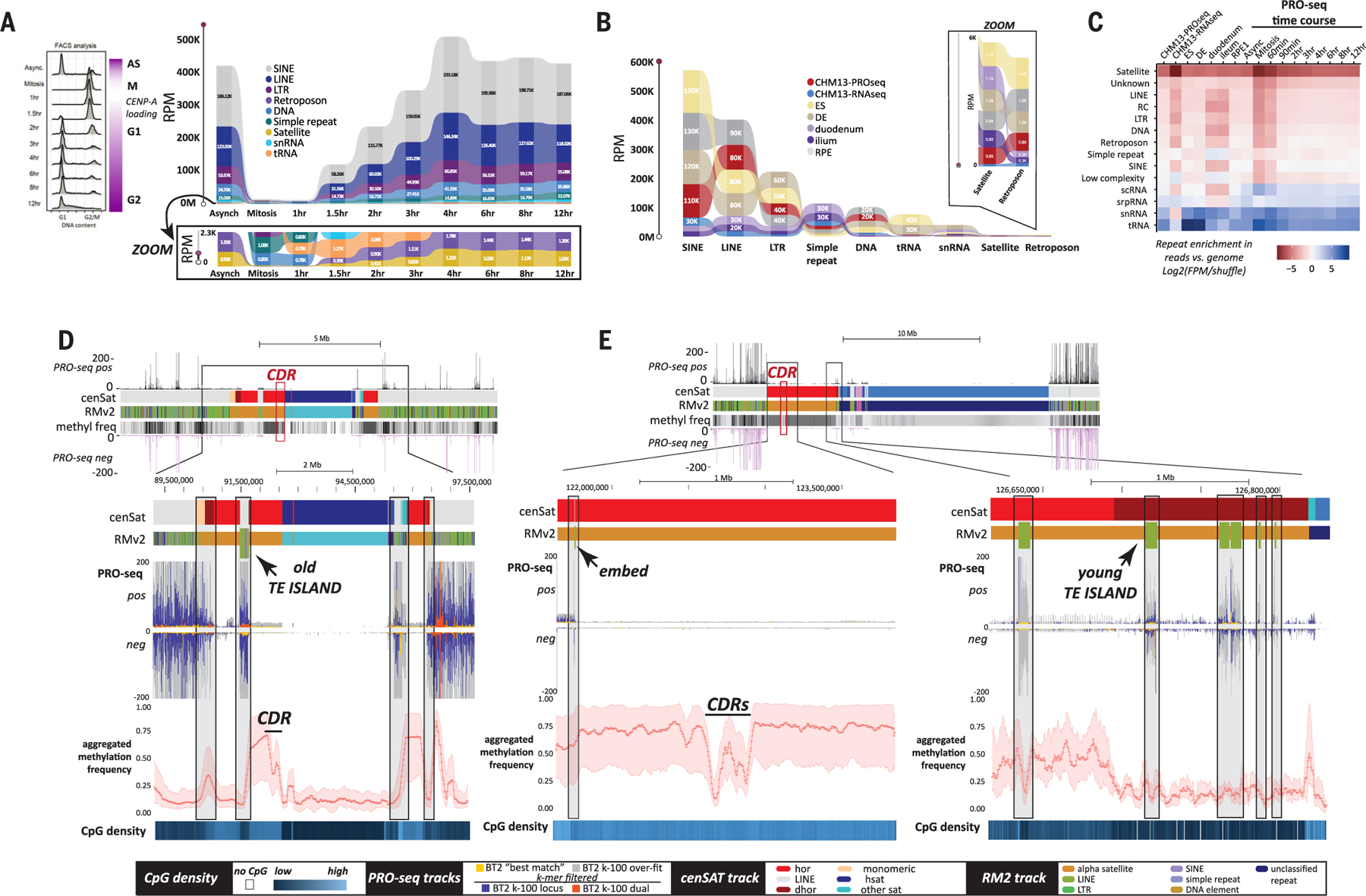
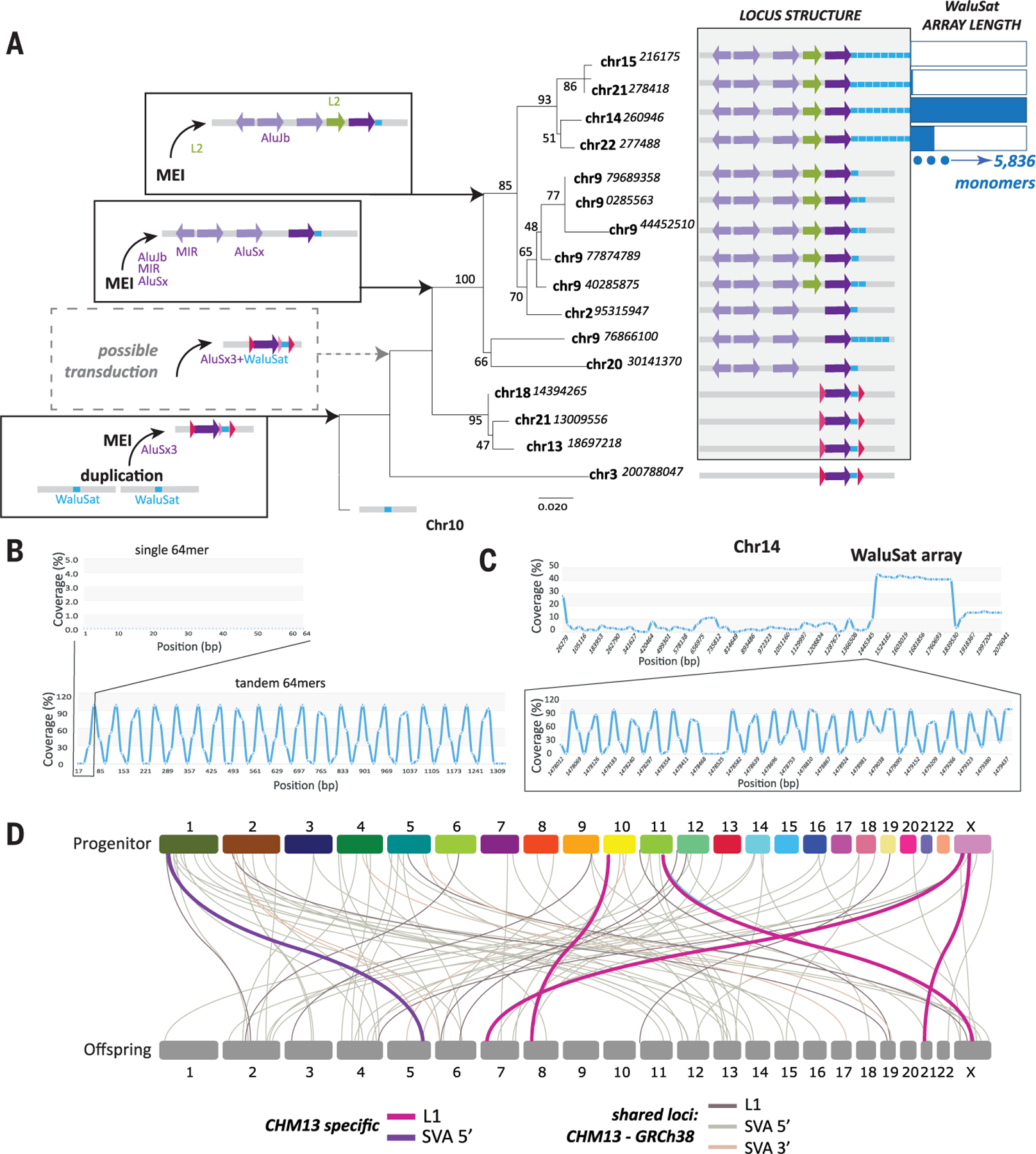
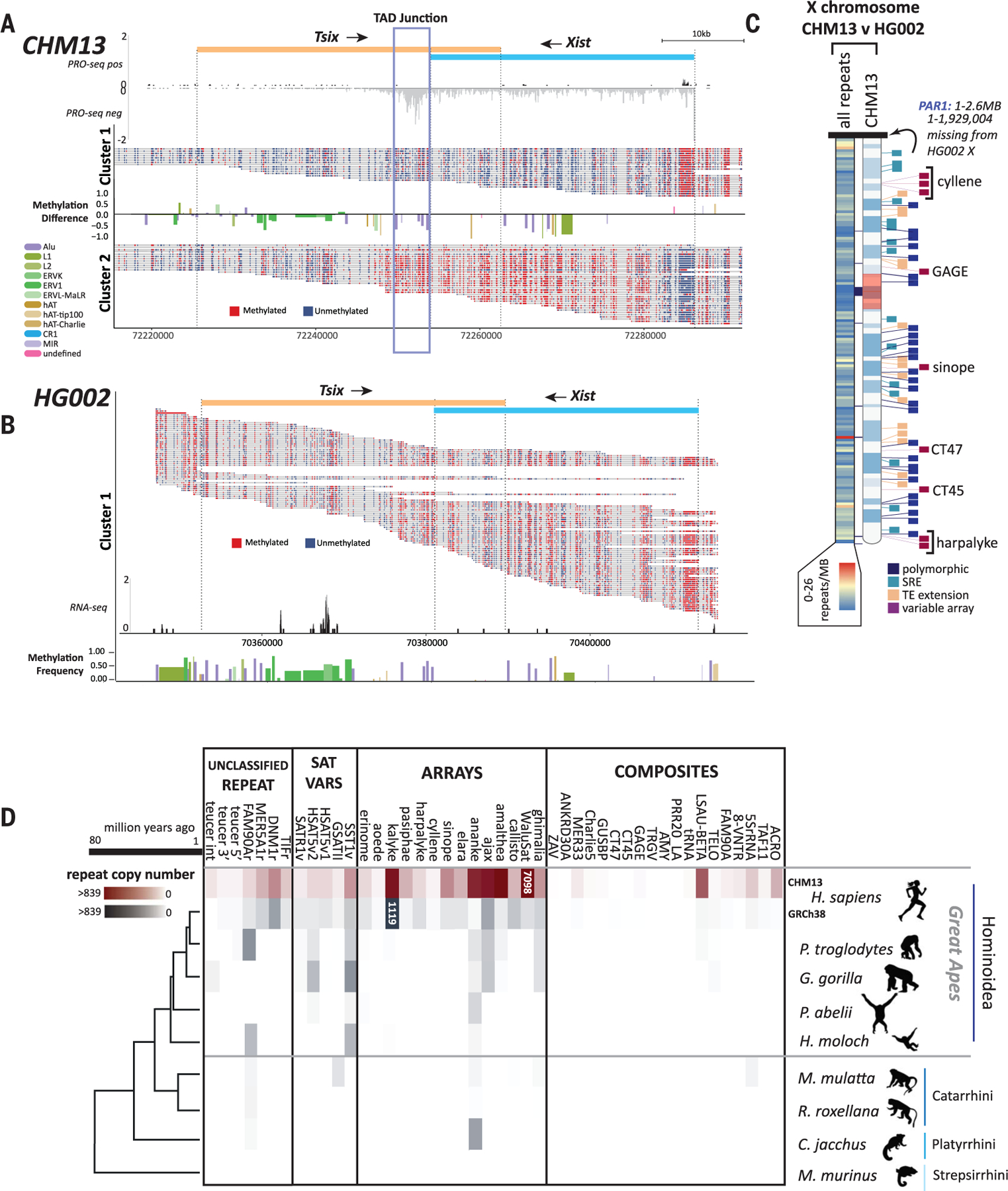
Mobile elements and repetitive genomic regions are sources of lineage-specific genomic innovation and uniquely fingerprint individual genomes. Comprehensive analyses of such repeat elements, including those found in more complex regions of the genome, require a complete, linear genome assembly. We present a de novo repeat discovery and annotation of the T2T-CHM13 human reference genome. We identified previously unknown satellite arrays, expanded the catalog of variants and families for repeats and mobile elements, characterized classes of complex composite repeats, and located retroelement transduction events. We detected nascent transcription and delineated CpG methylation profiles to define the structure of transcriptionally active retroelements in humans, including those in centromeres. These data expand our insight into the diversity, distribution, and evolution of repetitive regions that have shaped the human genome.
Conflict of interest statement
Figures
Comment in
-
The final pieces of the human genome.Nat Rev Genet. 2022 Jun;23(6):321. doi: 10.1038/s41576-022-00494-5. Nat Rev Genet. 2022. PMID: 35488041 No abstract available.
References
Publication types
MeSH terms
Grants and funding
- U24 HG010263/HG/NHGRI NIH HHS/United States
- R35 GM128857/GM/NIGMS NIH HHS/United States
- T32 GM007445/GM/NIGMS NIH HHS/United States
- R21 CA240199/CA/NCI NIH HHS/United States
- R01 HG010169/HG/NHGRI NIH HHS/United States
- U01 CA253481/CA/NCI NIH HHS/United States
- R01 HG002939/HG/NHGRI NIH HHS/United States
- U01 HG010971/HG/NHGRI NIH HHS/United States
- P20 GM103546/GM/NIGMS NIH HHS/United States
- R01 HG002385/HG/NHGRI NIH HHS/United States
- ZIA HG200398/ImNIH/Intramural NIH HHS/United States
- R01 GM132600/GM/NIGMS NIH HHS/United States
- R01 HG011274/HG/NHGRI NIH HHS/United States
- U24 HG006620/HG/NHGRI NIH HHS/United States
- U24 HG010136/HG/NHGRI NIH HHS/United States
- R21 HG010548/HG/NHGRI NIH HHS/United States
- R01 HG009909/HG/NHGRI NIH HHS/United States
- R01 GM123312/GM/NIGMS NIH HHS/United States
- R01 HG009190/HG/NHGRI NIH HHS/United States
- R24 DK106766/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
