

A complete reference genome improves analysis of human genetic variation
- PMID: 35357935
- PMCID: PMC9336181
- DOI: 10.1126/science.abl3533
A complete reference genome improves analysis of human genetic variation
Abstract
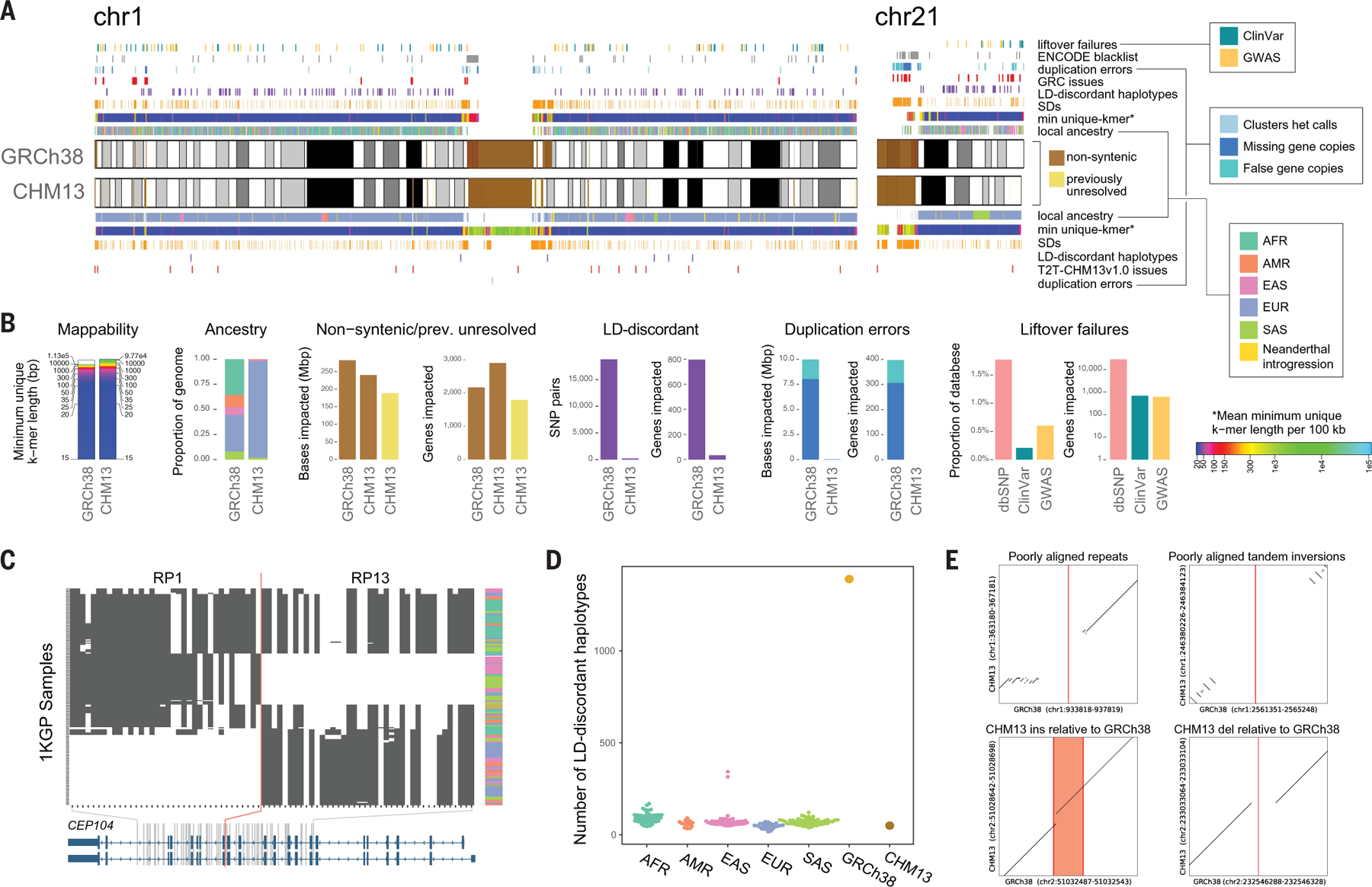
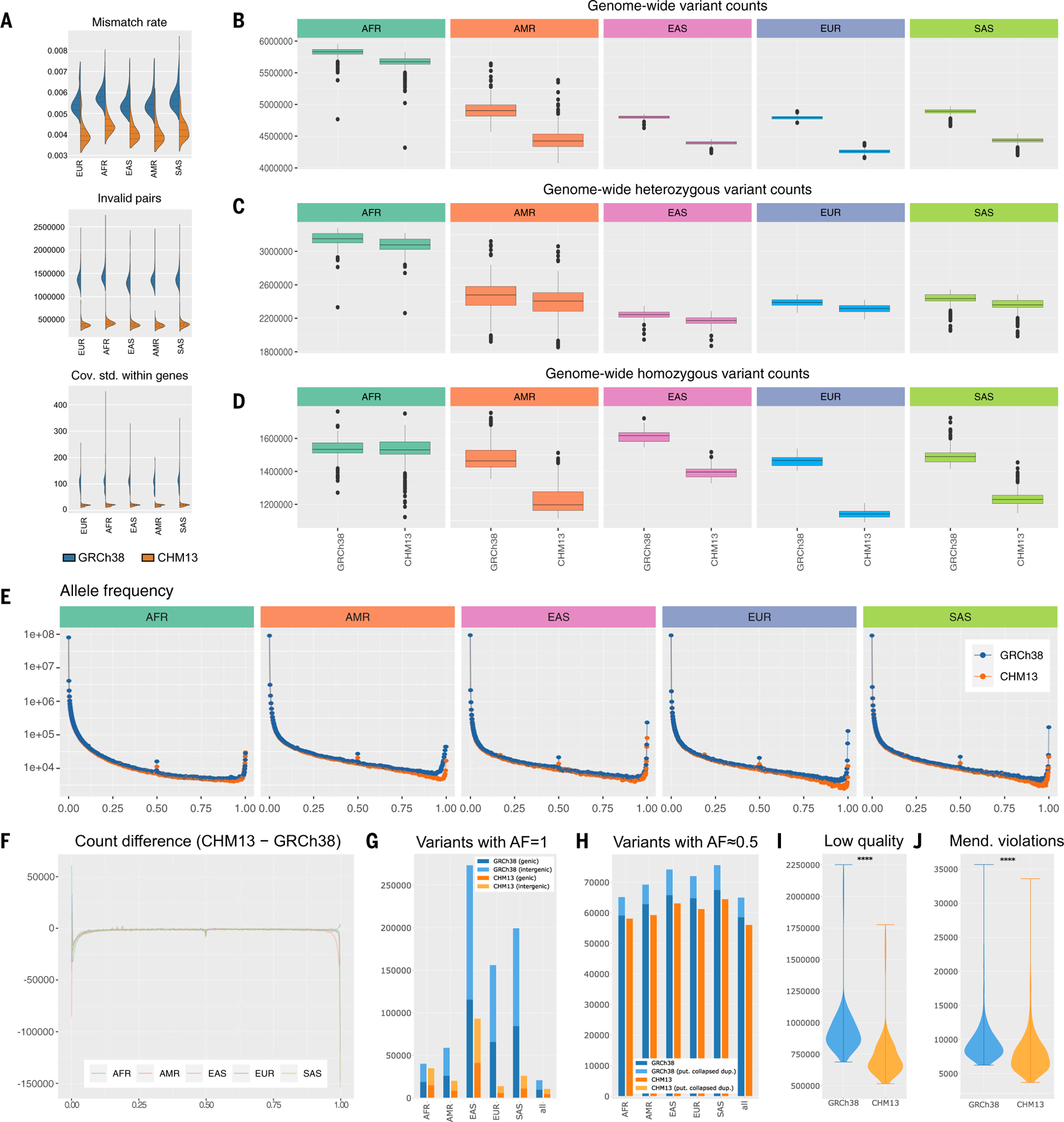
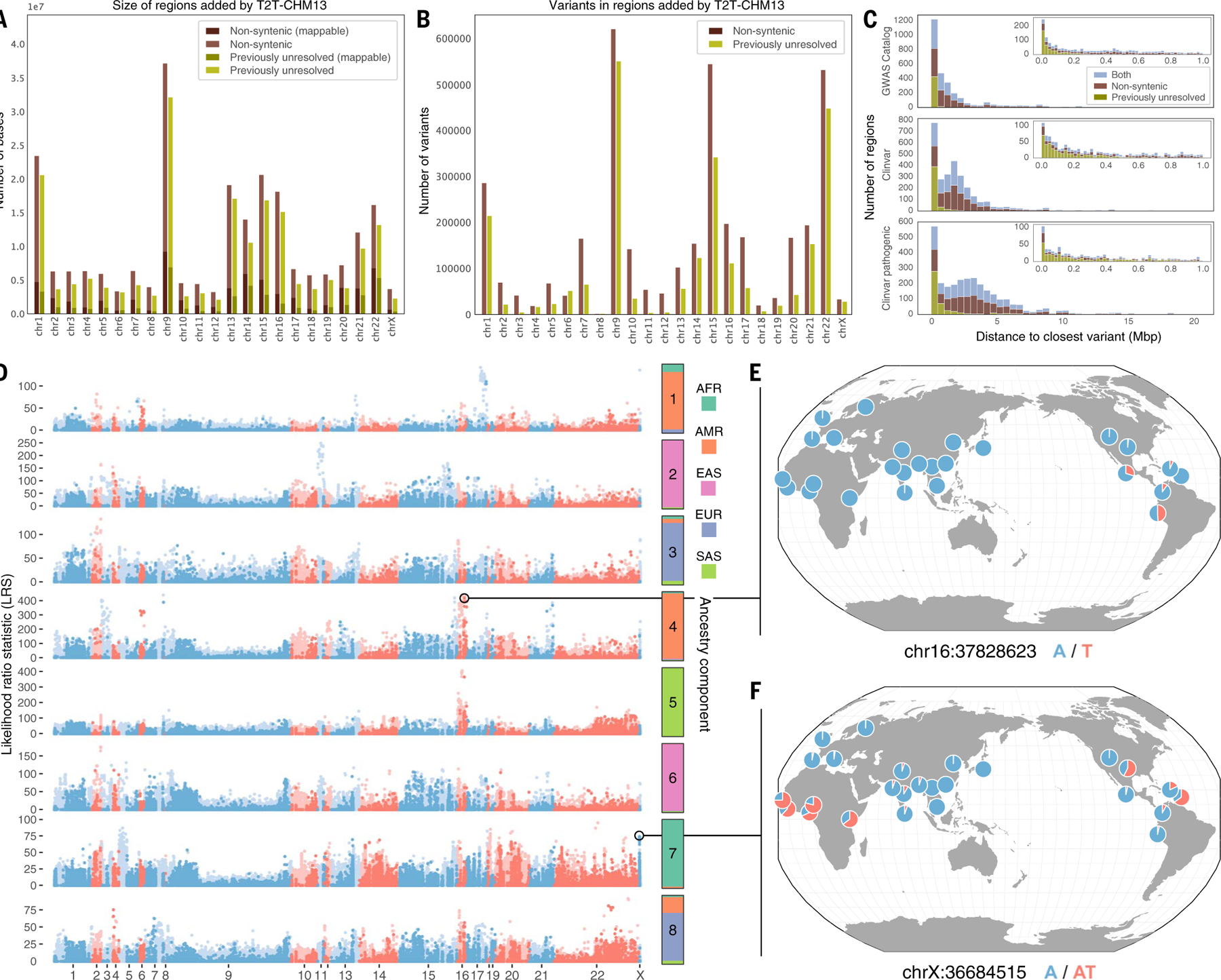
Compared to its predecessors, the Telomere-to-Telomere CHM13 genome adds nearly 200 million base pairs of sequence, corrects thousands of structural errors, and unlocks the most complex regions of the human genome for clinical and functional study. We show how this reference universally improves read mapping and variant calling for 3202 and 17 globally diverse samples sequenced with short and long reads, respectively. We identify hundreds of thousands of variants per sample in previously unresolved regions, showcasing the promise of the T2T-CHM13 reference for evolutionary and biomedical discovery. Simultaneously, this reference eliminates tens of thousands of spurious variants per sample, including reduction of false positives in 269 medically relevant genes by up to a factor of 12. Because of these improvements in variant discovery coupled with population and functional genomic resources, T2T-CHM13 is positioned to replace GRCh38 as the prevailing reference for human genetics.
Conflict of interest statement
Figures


Comment in
-
The final pieces of the human genome.Nat Rev Genet. 2022 Jun;23(6):321. doi: 10.1038/s41576-022-00494-5. Nat Rev Genet. 2022. PMID: 35488041 No abstract available.
References
Publication types
MeSH terms
Grants and funding
- U01 HG010961/HG/NHGRI NIH HHS/United States
- DP2 MH119424/MH/NIMH NIH HHS/United States
- U41 HG010972/HG/NHGRI NIH HHS/United States
- OT2 OD026682/OD/NIH HHS/United States
- R21 HG010548/HG/NHGRI NIH HHS/United States
- U01 HG010971/HG/NHGRI NIH HHS/United States
- U24 HG011853/HG/NHGRI NIH HHS/United States
- UM1 HG008898/HG/NHGRI NIH HHS/United States
- U24 HG010263/HG/NHGRI NIH HHS/United States
- R01 HG006677/HG/NHGRI NIH HHS/United States
- R35 GM133747/GM/NIGMS NIH HHS/United States
- R00 HG009532/HG/NHGRI NIH HHS/United States
- U01 CA253481/CA/NCI NIH HHS/United States
- R01 HG011274/HG/NHGRI NIH HHS/United States
- U24 HG006620/HG/NHGRI NIH HHS/United States
- R01 HG010485/HG/NHGRI NIH HHS/United States
- R24 DK106766/DK/NIDDK NIH HHS/United States
