

The immune microenvironment shapes transcriptional and genetic heterogeneity in chronic lymphocytic leukemia
- PMID: 35358998
- PMCID: PMC9811214
- DOI: 10.1182/bloodadvances.2021006941
The immune microenvironment shapes transcriptional and genetic heterogeneity in chronic lymphocytic leukemia
Abstract
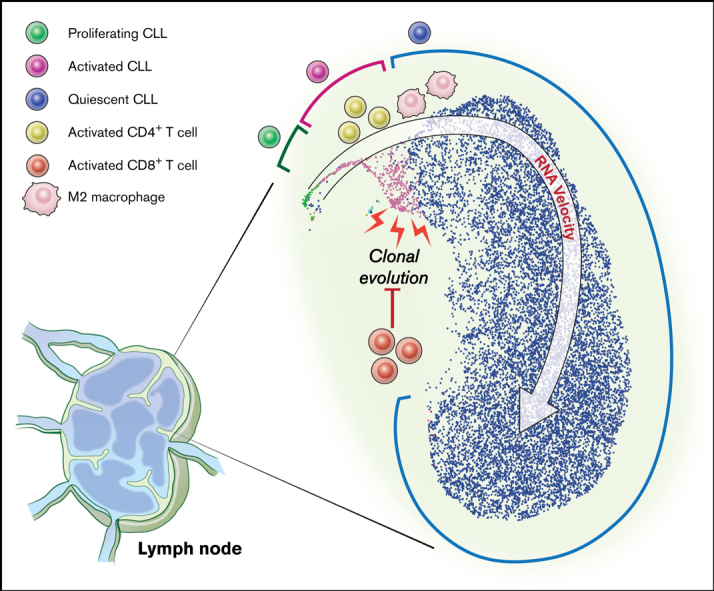
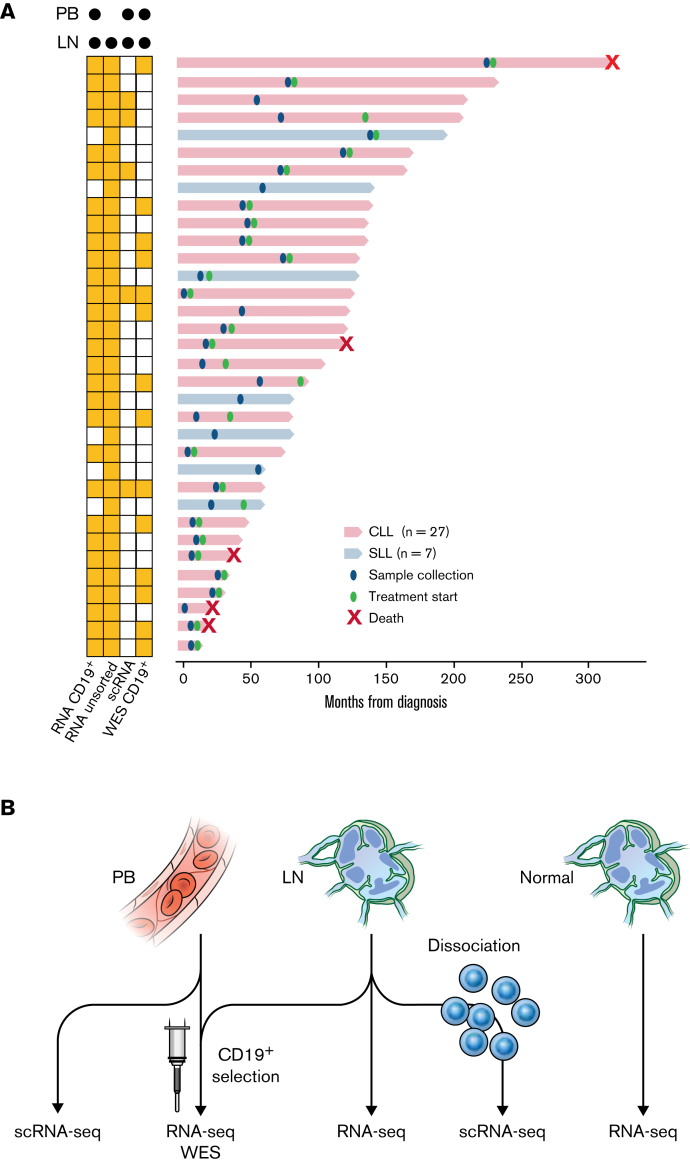
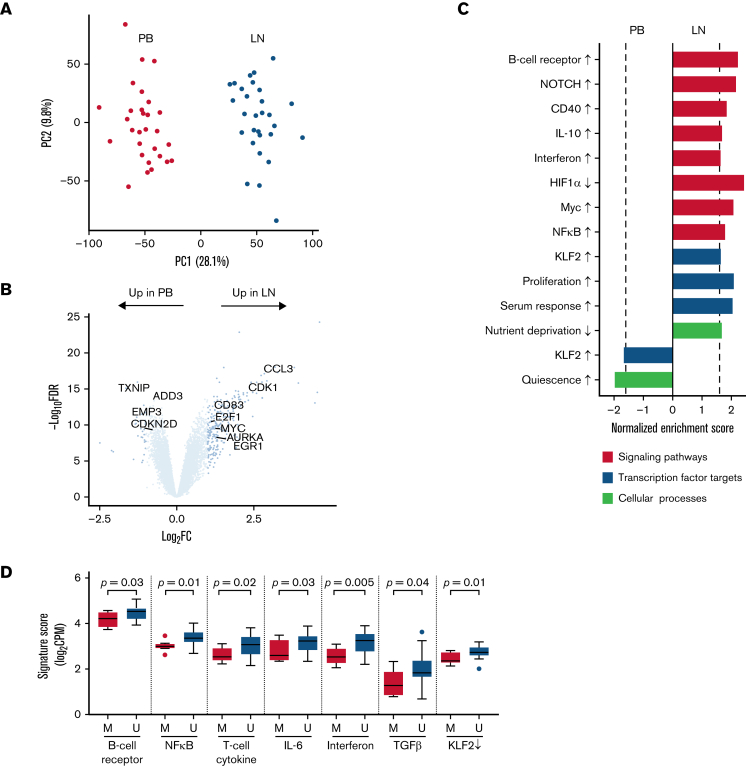
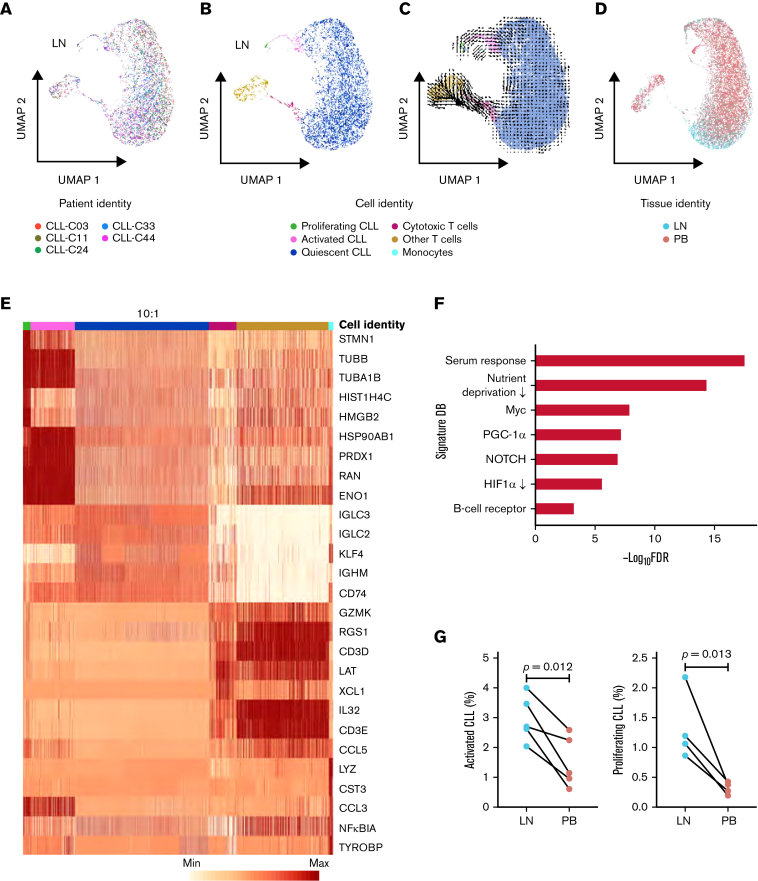
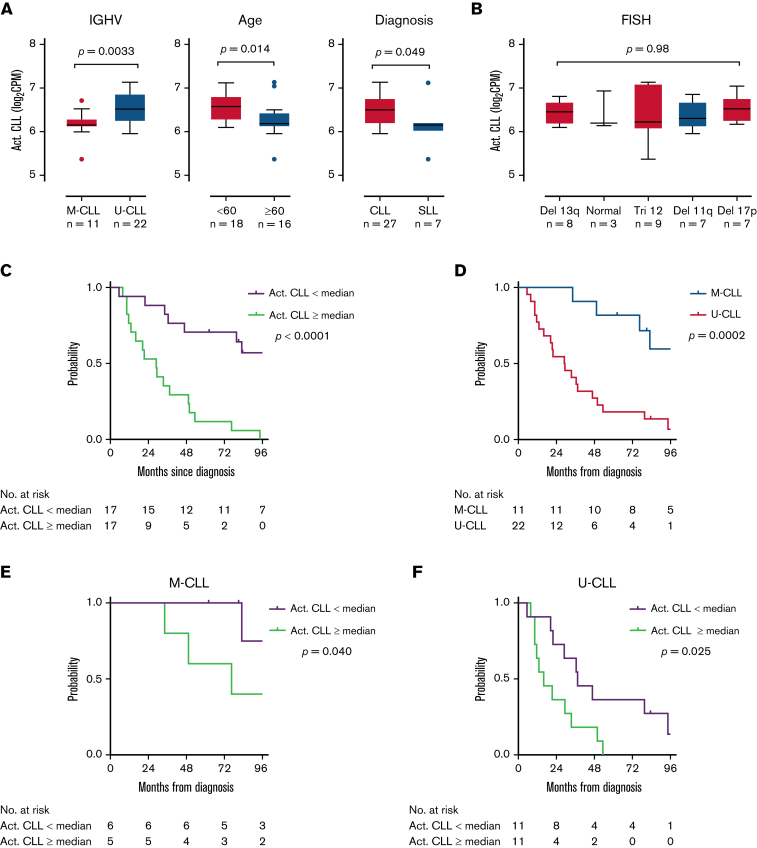
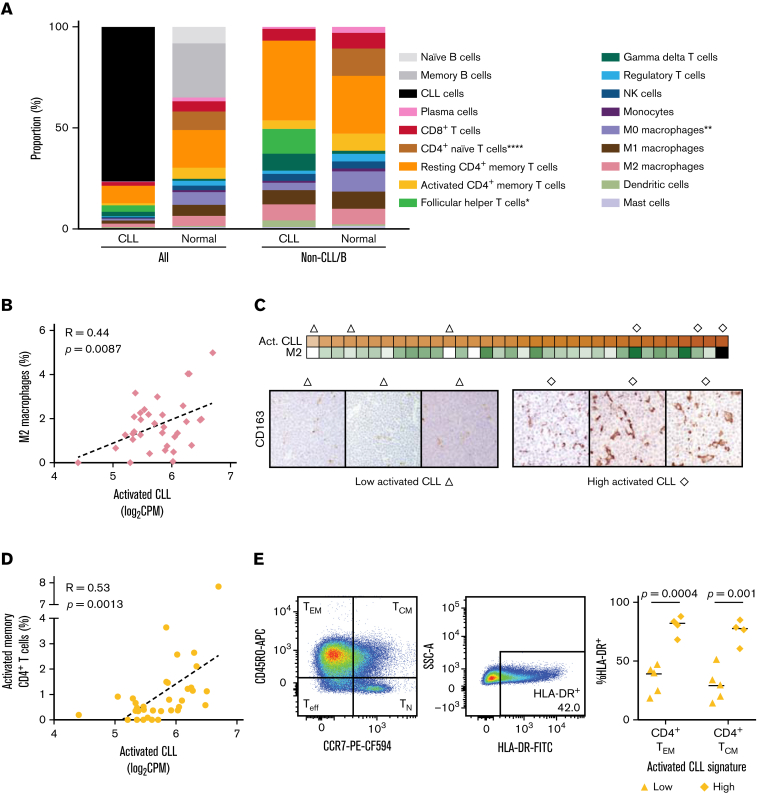
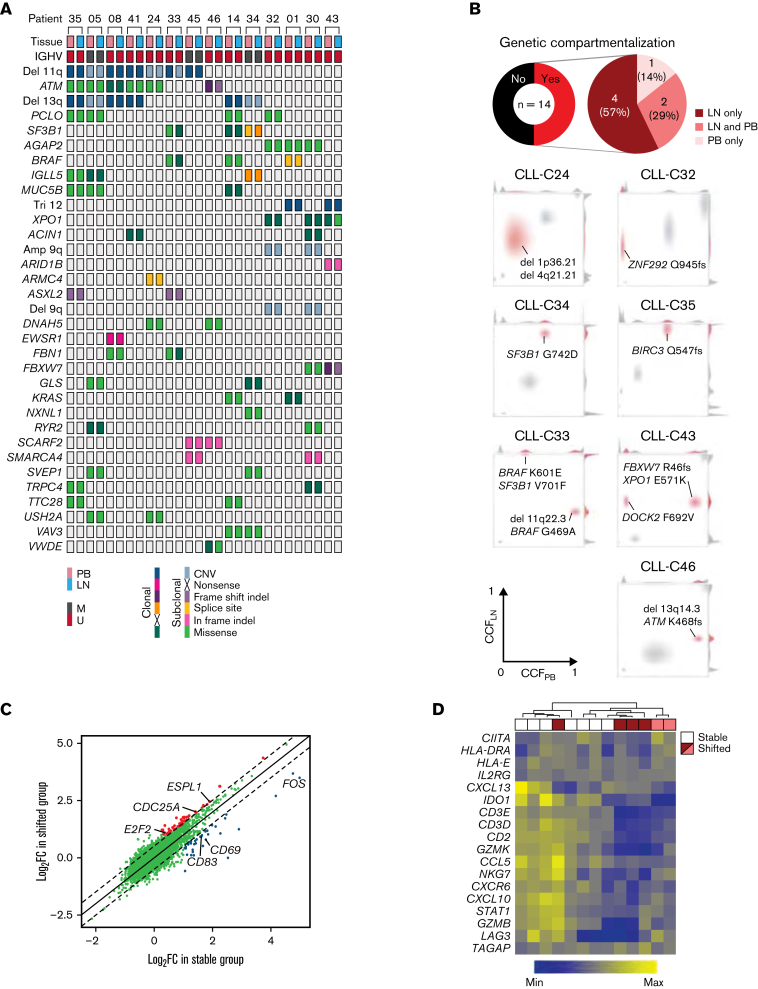
In chronic lymphocytic leukemia (CLL), B-cell receptor signaling, tumor-microenvironment interactions, and somatic mutations drive disease progression. To better understand the intersection between the microenvironment and molecular events in CLL pathogenesis, we integrated bulk transcriptome profiling of paired peripheral blood (PB) and lymph node (LN) samples from 34 patients. Oncogenic processes were upregulated in LN compared with PB and in immunoglobulin heavy-chain variable (IGHV) region unmutated compared with mutated cases. Single-cell RNA sequencing (scRNA-seq) distinguished 3 major cell states: quiescent, activated, and proliferating. The activated subpopulation comprised only 2.2% to 4.3% of the total tumor bulk in LN samples. RNA velocity analysis found that CLL cell fate in LN is unidirectional, starts in the proliferating state, transitions to the activated state, and ends in the quiescent state. A 10-gene signature derived from activated tumor cells was associated with inferior treatment-free survival (TFS) and positively correlated with the proportion of activated CD4+ memory T cells and M2 macrophages in LN. Whole exome sequencing (WES) of paired PB and LN samples showed subclonal expansion in LN in approximately half of the patients. Since mouse models have implicated activation-induced cytidine deaminase in mutagenesis, we compared AICDA expression between cases with and without clonal evolution but did not find a difference. In contrast, the presence of a T-cell inflamed microenvironment in LN was associated with clonal stability. In summary, a distinct minor tumor subpopulation underlies CLL pathogenesis and drives the clinical outcome. Clonal trajectories are shaped by the LN milieu, where T-cell immunity may contribute to suppressing clonal outgrowth. The clinical study is registered at clinicaltrials.gov as NCT00923507.
Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: C.S. receives research support from Genmab. M.J.B. is an employee at AstraZeneca. S.H.G. is a consultant for Novalgen Ltd and holds patents related to ROR1 therapies; has received honoraria, conference support, and speakers fees from AstraZeneca, AbbVie, and Janssen. N.S.S. is a consultant for and has received speaker fees from AbbVie, Pharmacyclics LLC, an AbbVie company, and Janssen; and is a member of the advisory board for TG Therapeutics, Innocare, BeiGene, KyowaKirin, ADC therapeutics, and Kite. G.G. receives research funds from IBM and Pharmacyclics; and is an inventor on patent applications related to MSMuTect, MSMutSig, MSIDetect, POLYSOLVER, and SignatureAnalyzer-GPU; is a founder, consultant, and holds privately held equity in Scorpion Therapeutics. C.J.W. holds equity in BioNTech, Inc and receives research funding from Pharmacyclics. A.W. receives research support from Pharmacyclics LLC, an AbbVie Company, Acerta LLC, a member of the AstraZeneca Group, Merck, Verastem, Nurix, and Genmab. The remaining authors declare no competing financial interests.
Figures
References
-
- Granziero L, Ghia P, Circosta P, et al. Survivin is expressed on CD40 stimulation and interfaces proliferation and apoptosis in B-cell chronic lymphocytic leukemia. Blood. 2001;97(9):2777–2783. - PubMed
-
- Aguilar-Hernandez MM, Blunt MD, Dobson R, et al. IL-4 enhances expression and function of surface IgM in CLL cells. Blood. 2016;127(24):3015–3025. - PubMed
-
- Ghia P, Strola G, Granziero L, et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4+, CD40L+ T cells by producing CCL22. Eur J Immunol. 2002;32(5):1403–1413. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
