

Autoimmune pulmonary alveolar proteinosis exacerbated by steroid therapy due to misdiagnosis as anti-aminoacyl-tRNA synthetase (ARS) antibody positive- interstitial pneumonia: a case report
- PMID: 35361191
- PMCID: PMC8973538
- DOI: 10.1186/s12890-022-01909-z
Autoimmune pulmonary alveolar proteinosis exacerbated by steroid therapy due to misdiagnosis as anti-aminoacyl-tRNA synthetase (ARS) antibody positive- interstitial pneumonia: a case report
Abstract
Background: Anti-aminoacyl-tRNA synthetase (anti-ARS) antibodies are myositis-specific autoantibodies that have been identified in a subset of patients with interstitial pneumonia who do not present with dermatomyositis or polymyositis. Anti-ARS antibody-positive interstitial pneumonia is commonly treated with steroids or immunosuppressive agents and is usually responsive to these therapies. Here, we present in detail a case in which respiratory failure of a patient diagnosed with anti-ARS antibody-positive interstitial pneumonia was exacerbated by treatment with steroids and immunosuppressive agents. Further examination revealed misdiagnosis of this patient and a subsequent diagnosis of autoimmune pulmonary alveolar proteinosis.
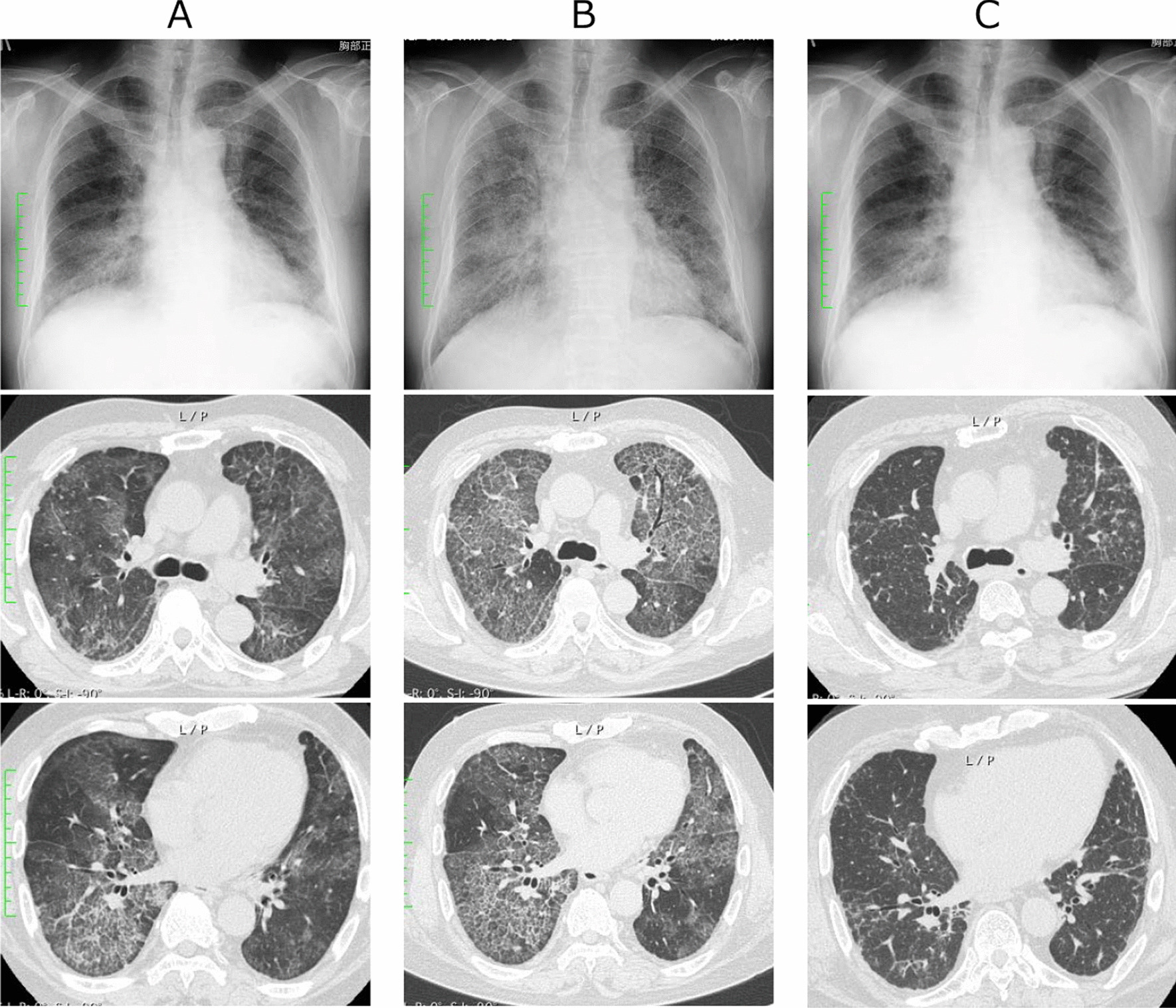
Case presentation: A 66-year-old man presented to the hospital with dyspnea on exertion, which resulted in the detection of interstitial pneumonia. Serum anti-ARS antibodies were detected; however, there were no other findings suggestive of myositis. Pulmonary alveolar proteinosis (PAP) was suspected based on the marked increase in serum KL-6 and chest computed tomography findings. The bronchoalveolar lavage revealed no milky changes in the lavage fluid. After treatment with steroids and initiation of immunosuppressive agents for anti-ARS antibody-positive interstitial pneumonia, respiratory failure and chest imaging findings showed worsening of the condition. Bronchoscopy was repeated, and milk-like alveolar lavage fluid was collected; serum anti-granulocyte macrophage colony-stimulating factor antibody was identified. Steroids and immunosuppressive agents were gradually tapered and discontinued, and the patient's condition stabilized after repeated alveolar lavage under general anesthesia.
Conclusion: Due to similar presentation, PAP can be misdiagnosed as interstitial pneumonia. If pulmonary lesions due to interstitial pneumonia are exacerbated by immunosuppressive treatment, physicians should reconsider the diagnosis and include PAP in the differential diagnosis.
Keywords: Anti-ARS antibody; Anti-PL-7 antibody; Autoimmune alveolar proteinosis; Case report; Steroid.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Uchida K, Nakata K, Trapnell BC, Terakawa T, Hamano E, Mikami A, et al. High-affinity autoantibodies specifically eliminate granulocyte-macrophage colony-stimulating factor activity in the lungs of patients with idiopathic pulmonary alveolar proteinosis. Blood. 2004;103:1089–1098. doi: 10.1182/blood-2003-05-1565. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
