

Chasing perfection: validation and polishing strategies for telomere-to-telomere genome assemblies
- PMID: 35361931
- PMCID: PMC9812399
- DOI: 10.1038/s41592-022-01440-3
Chasing perfection: validation and polishing strategies for telomere-to-telomere genome assemblies
Abstract
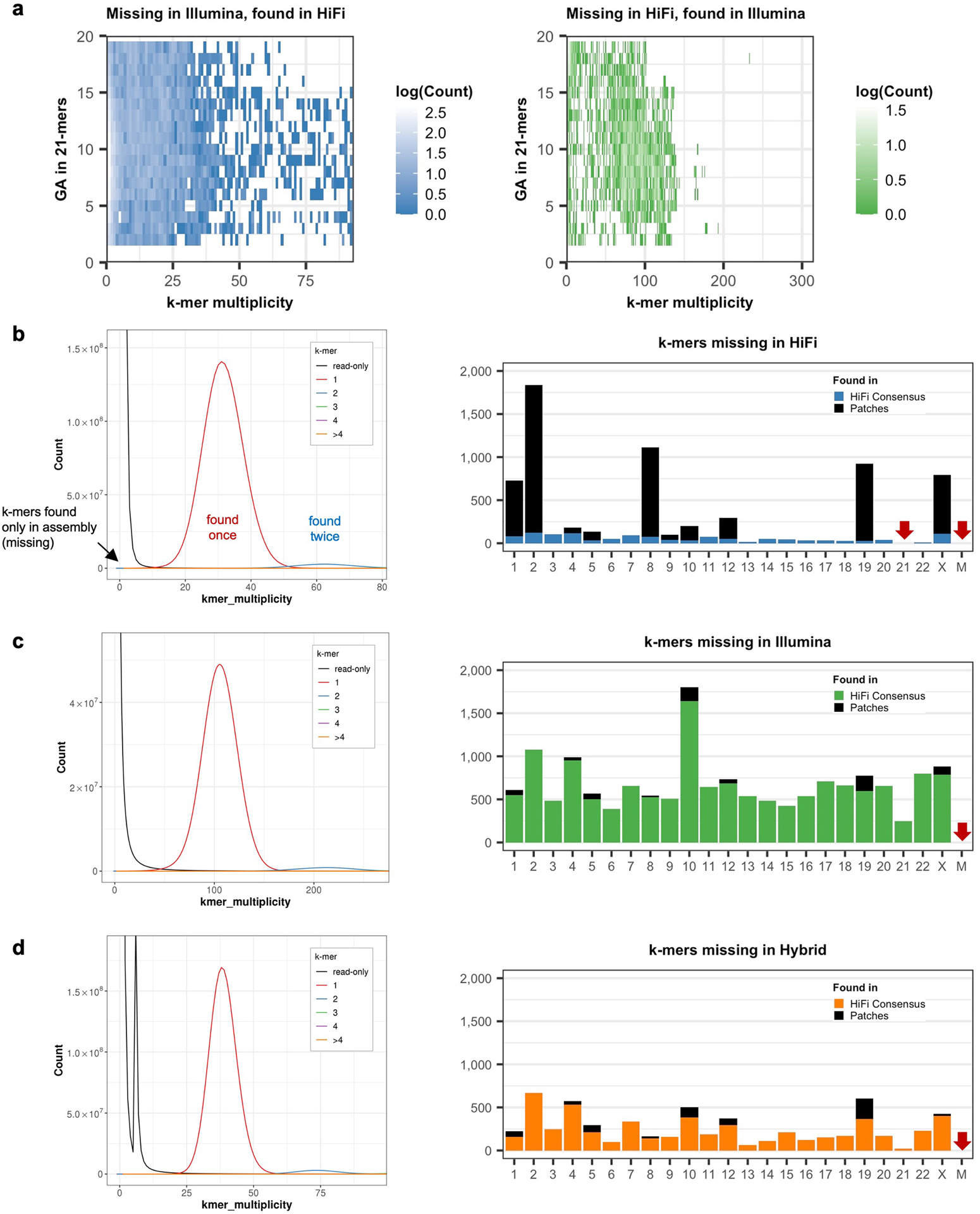
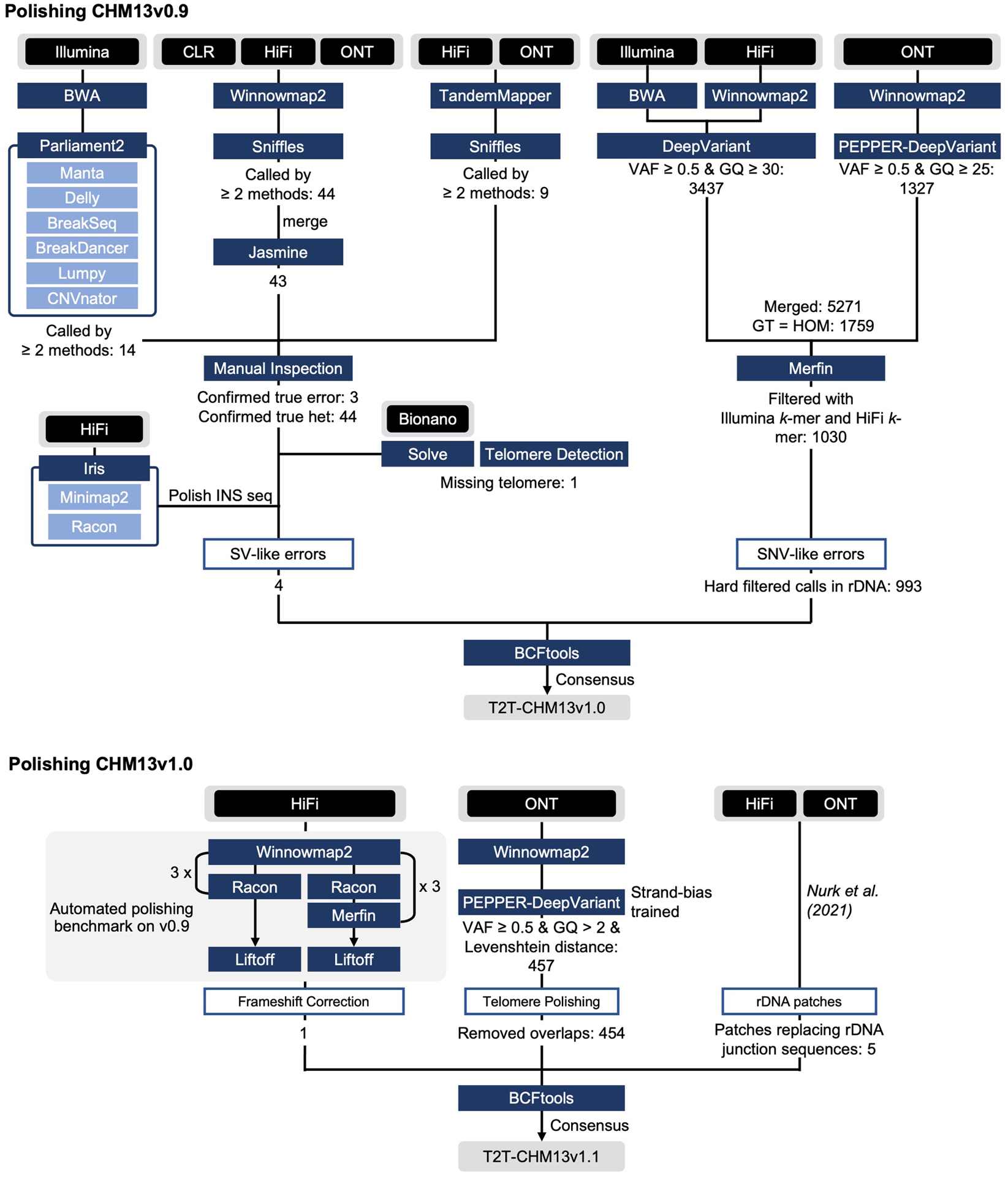
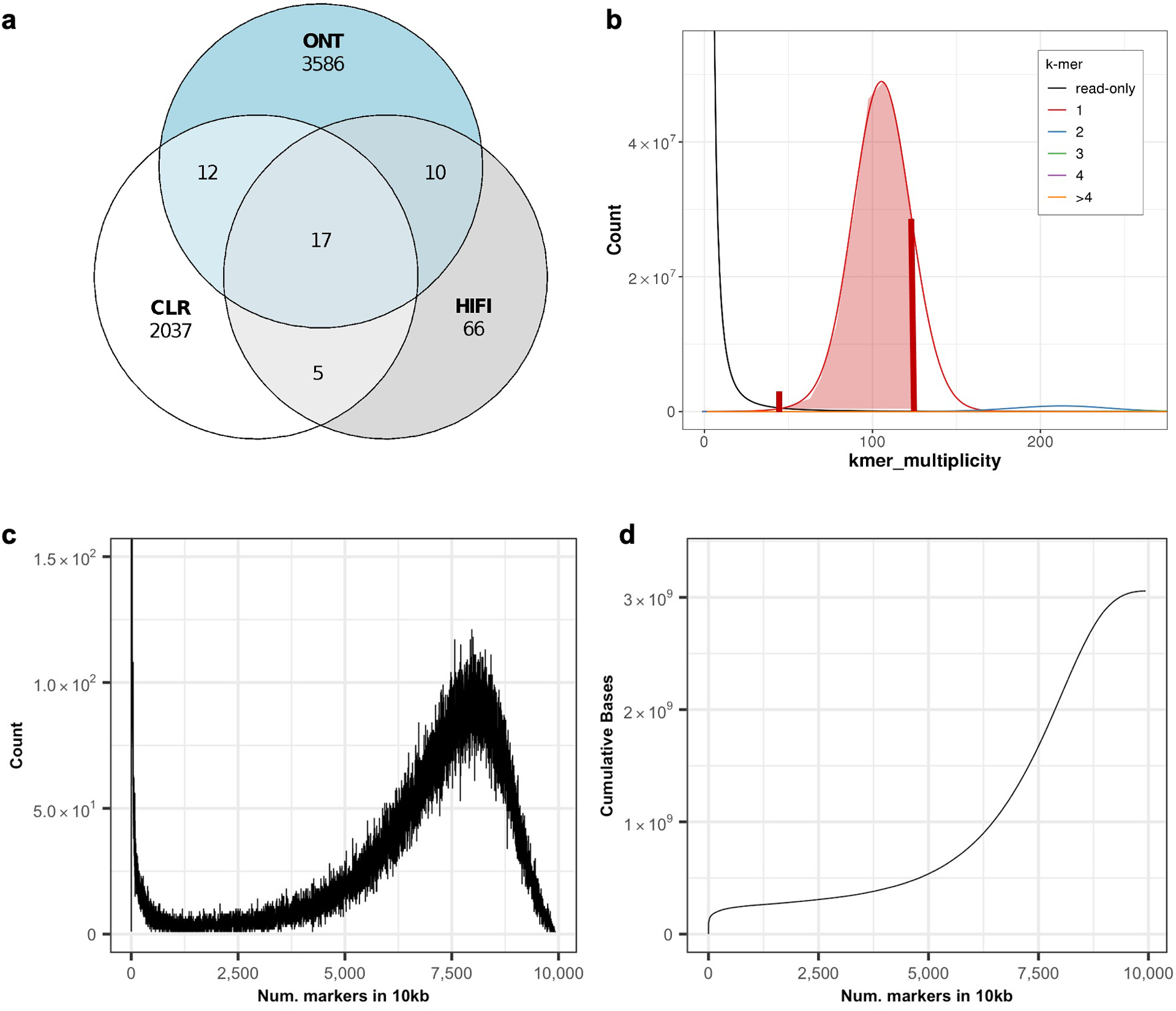
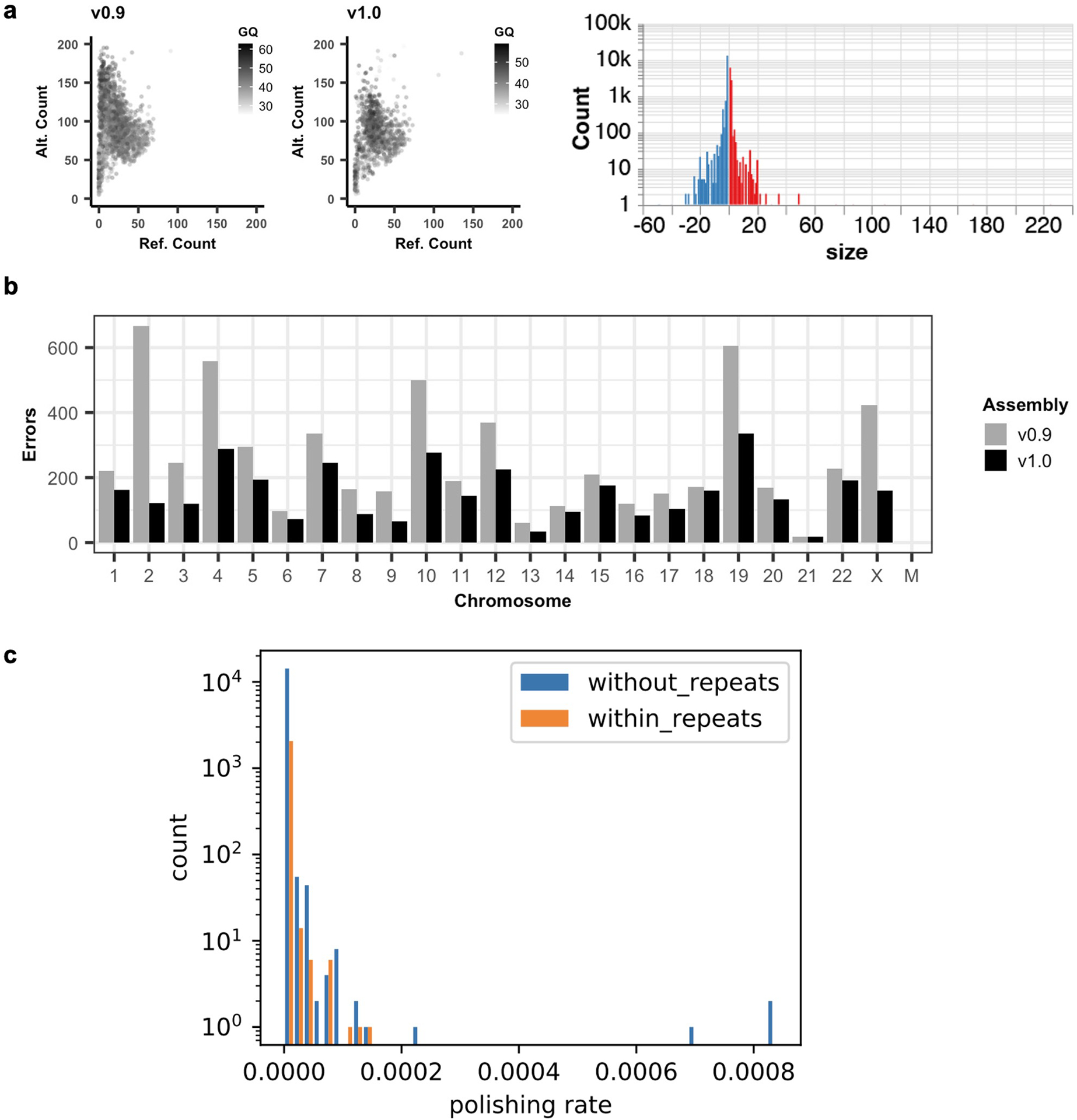
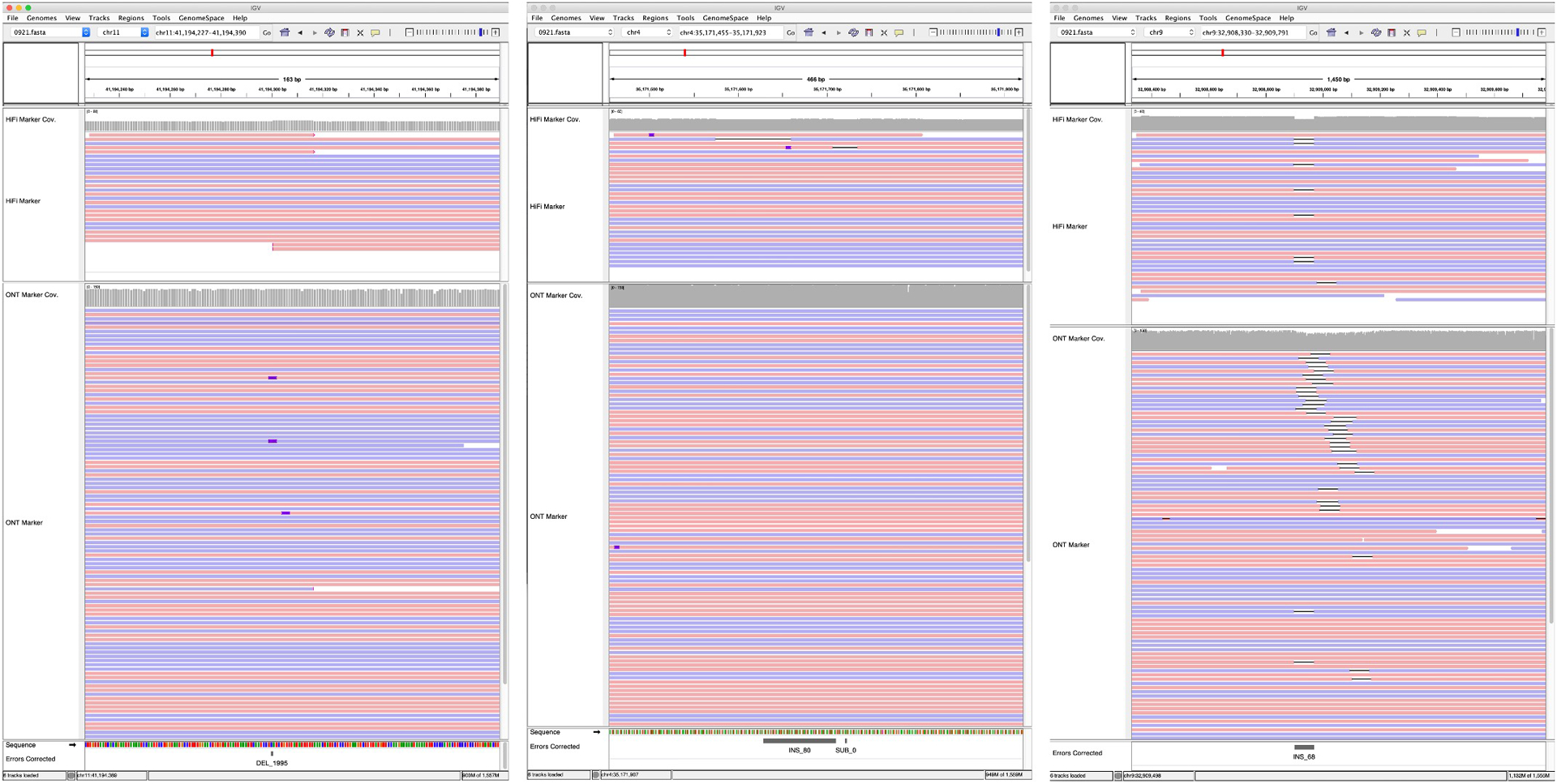
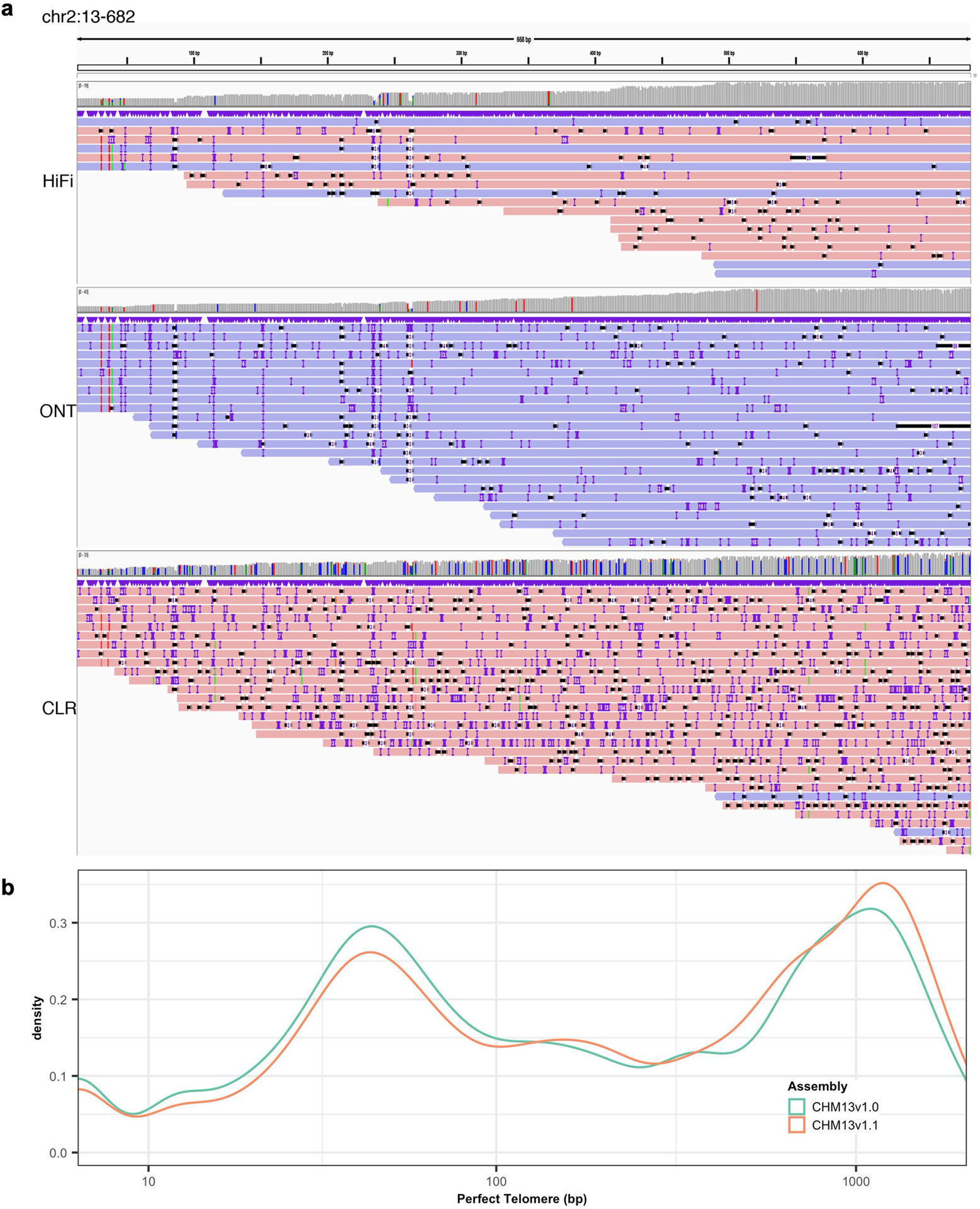
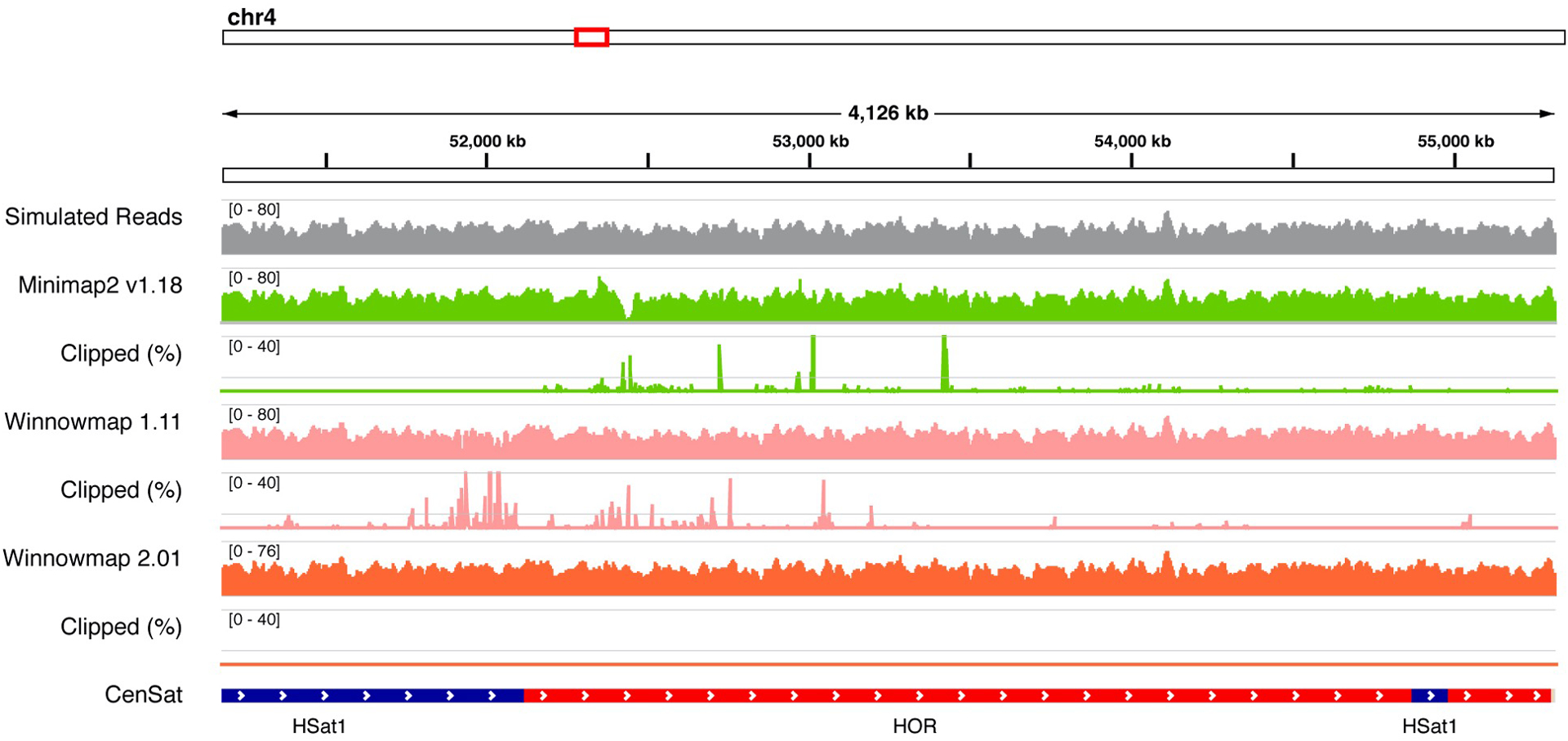
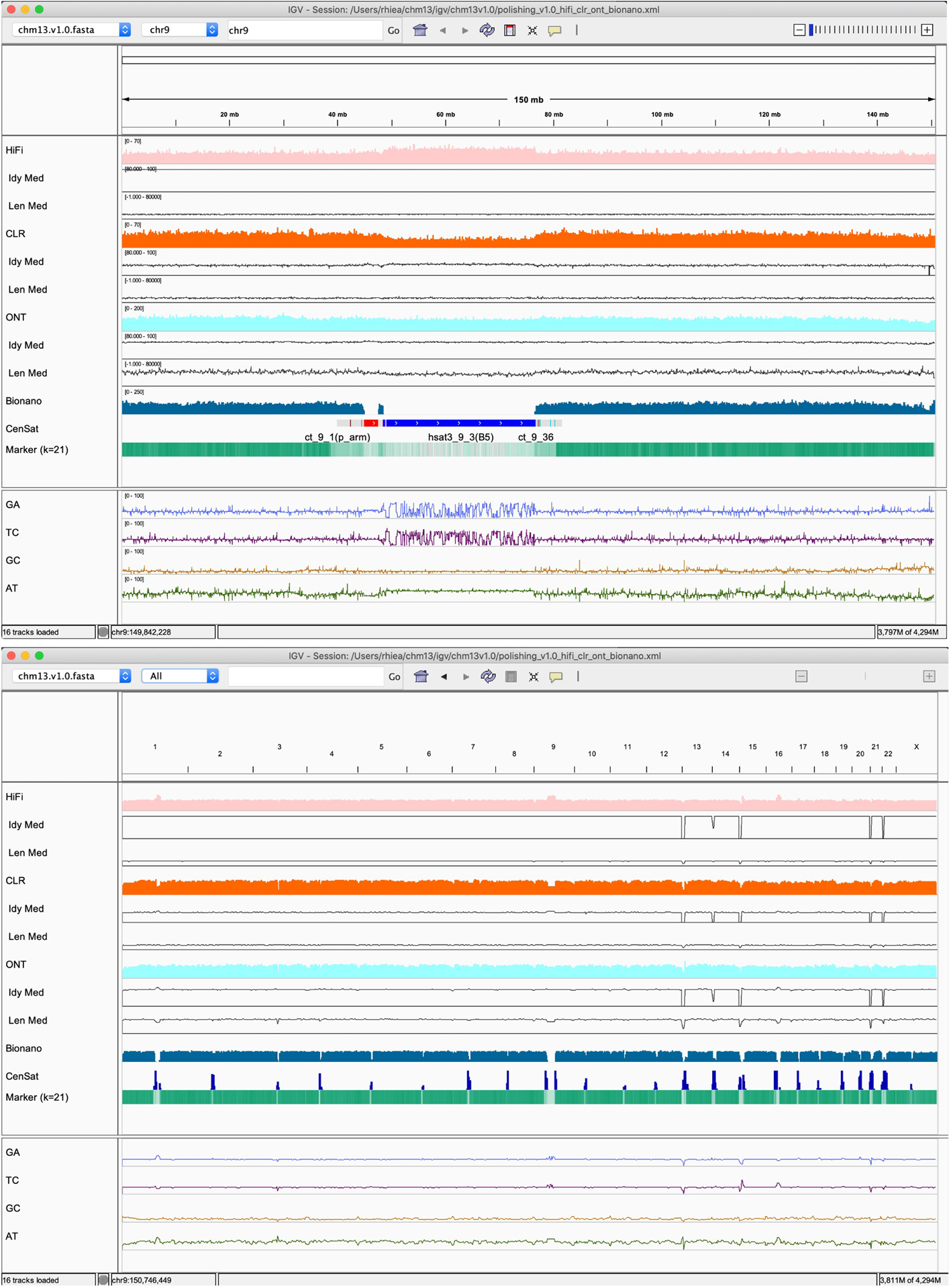
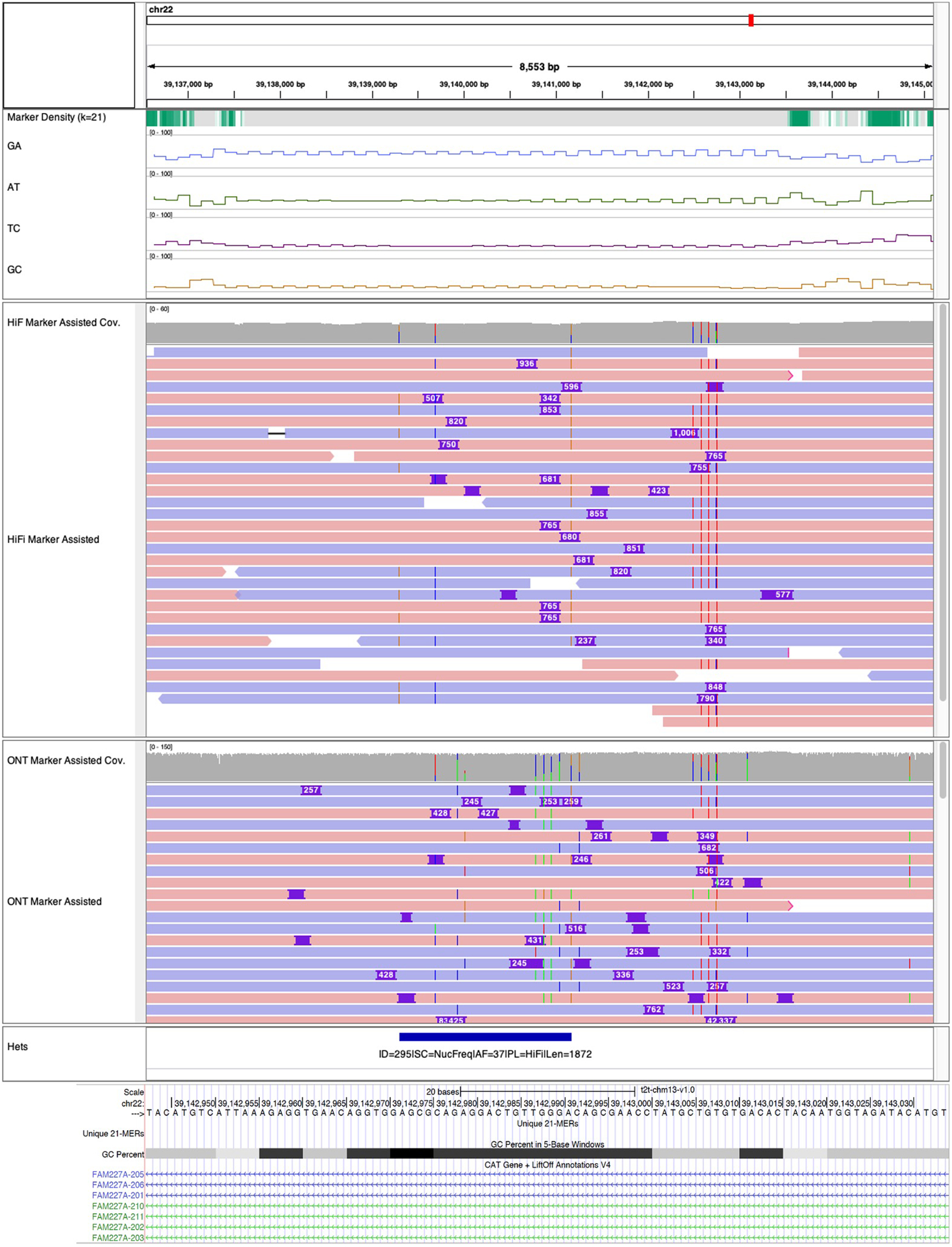
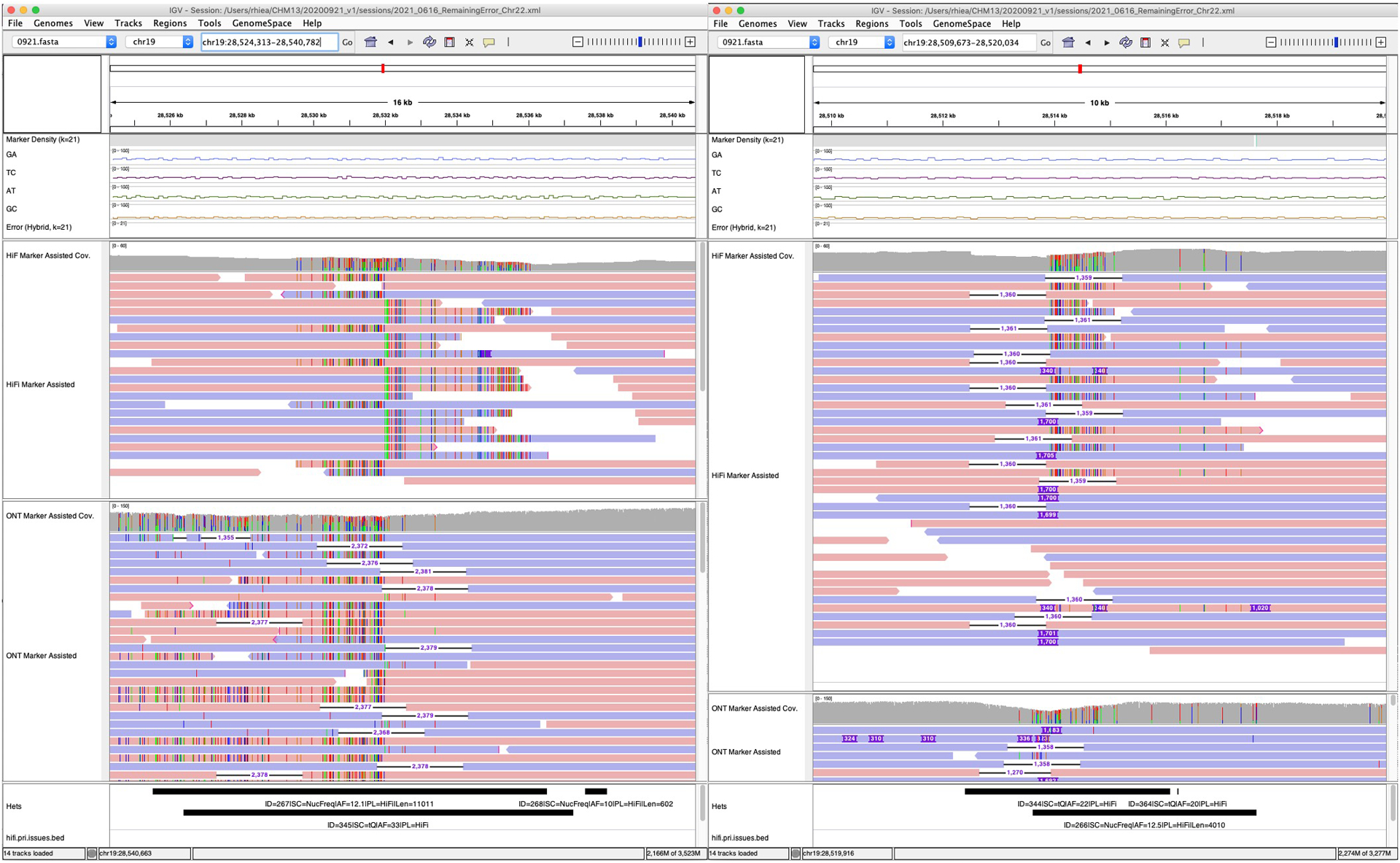
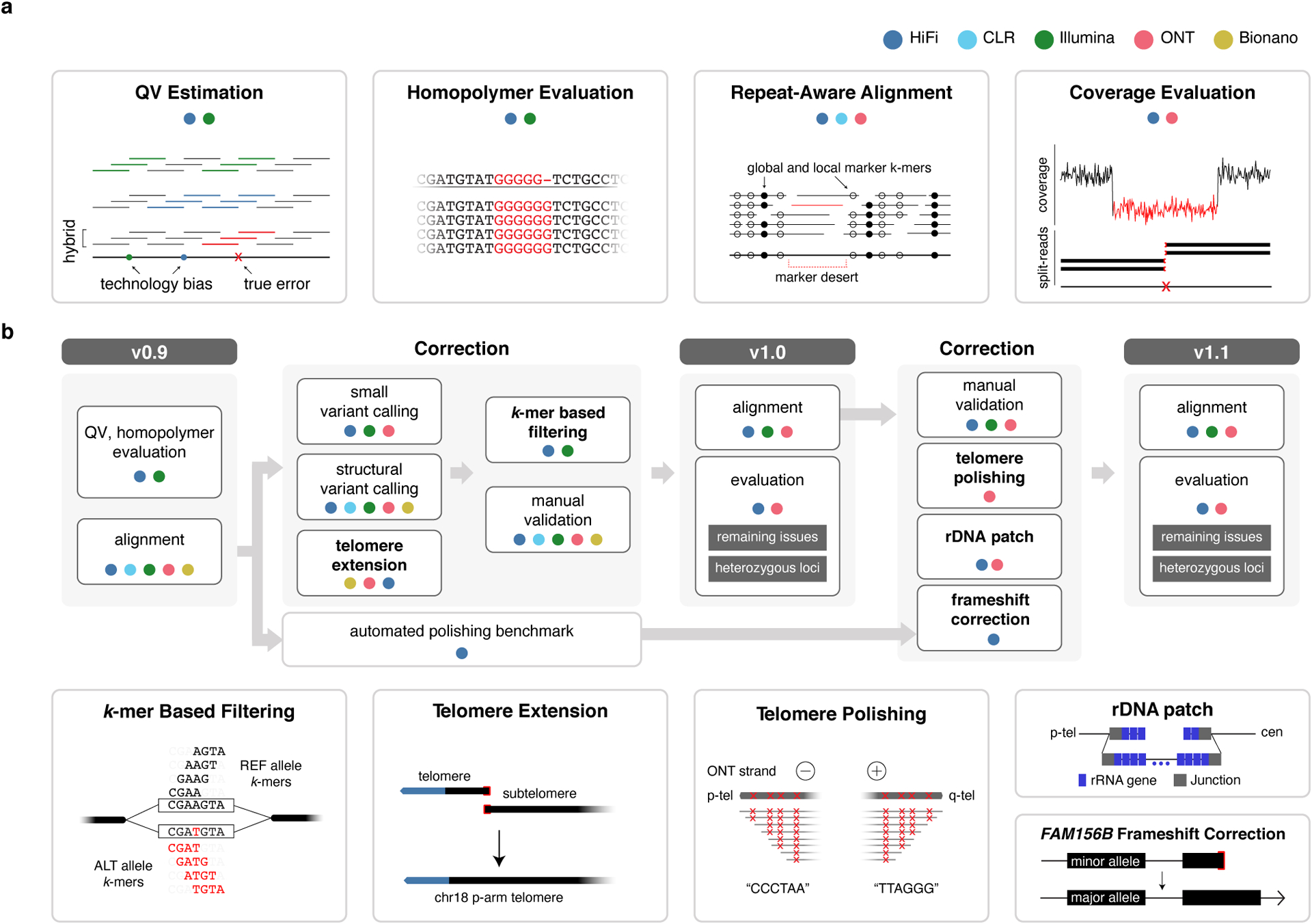
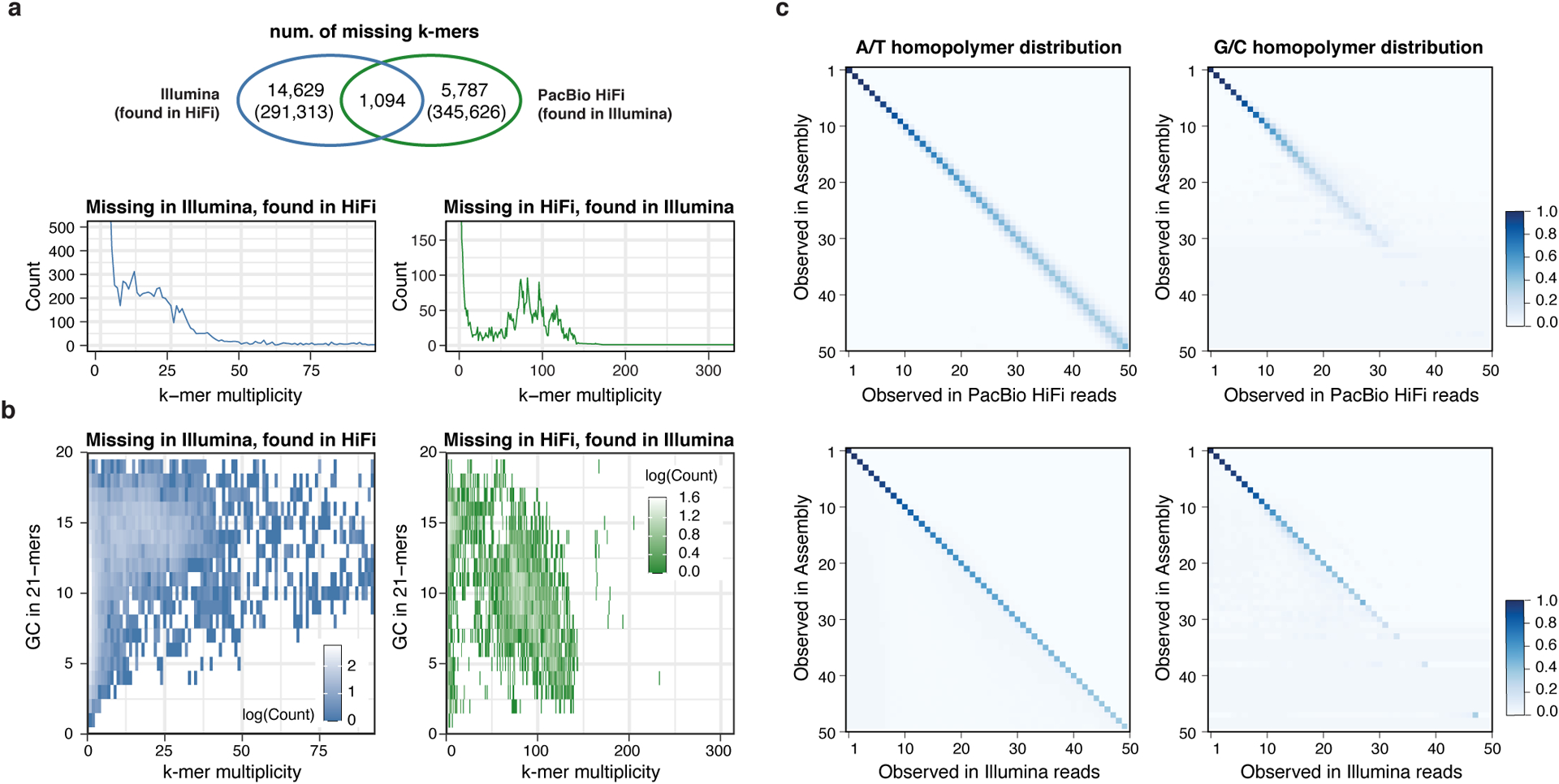
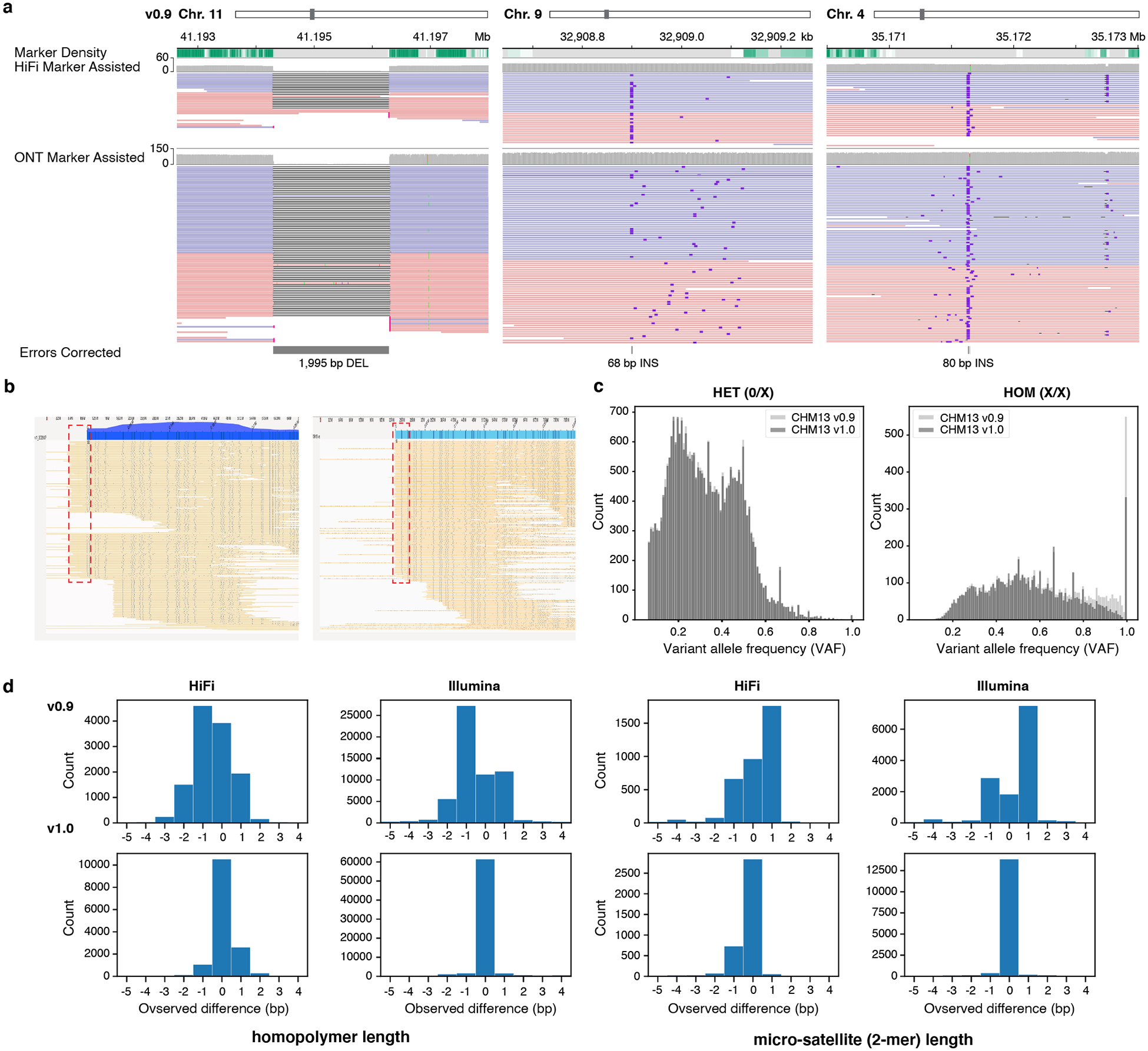
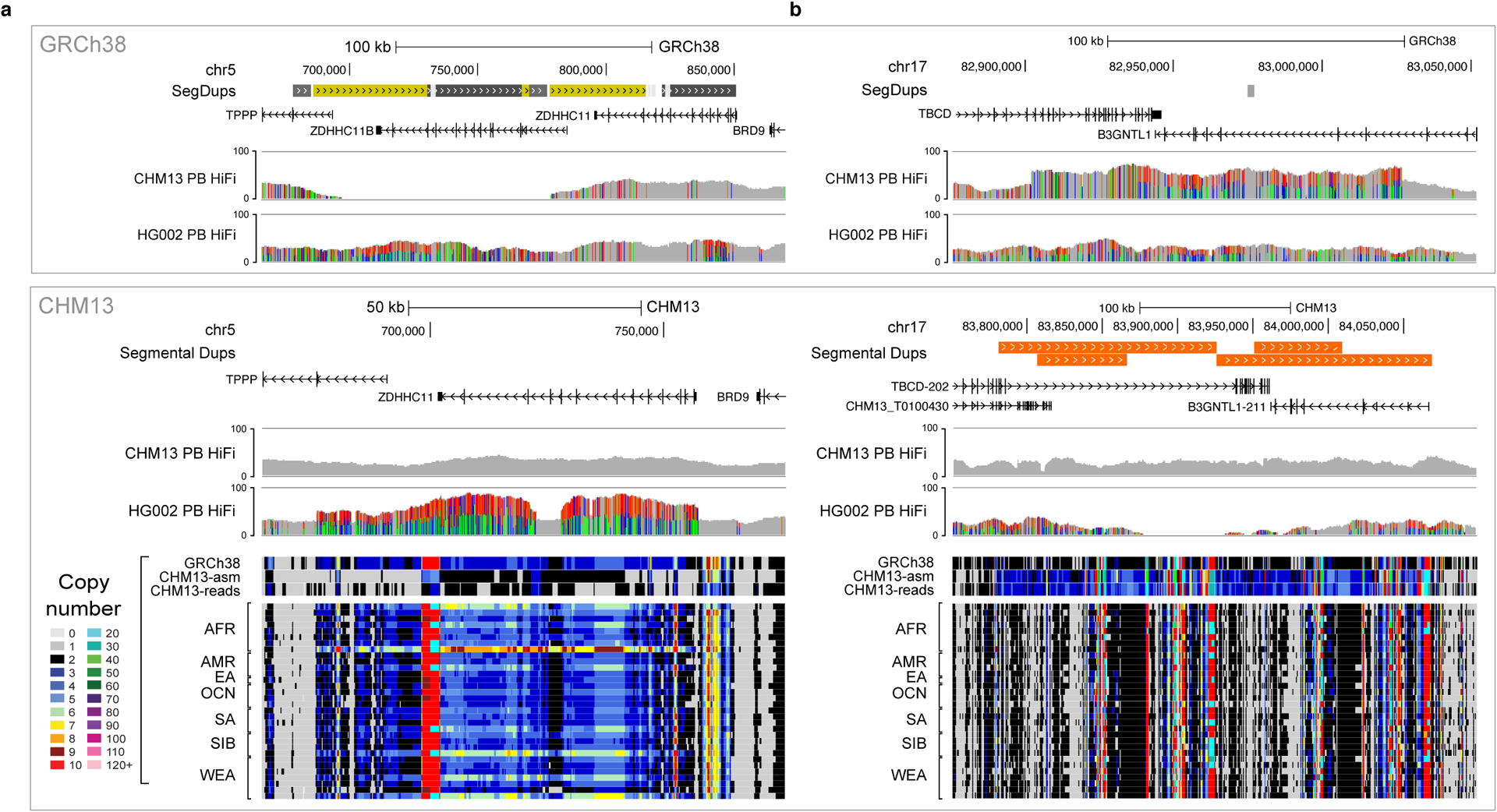
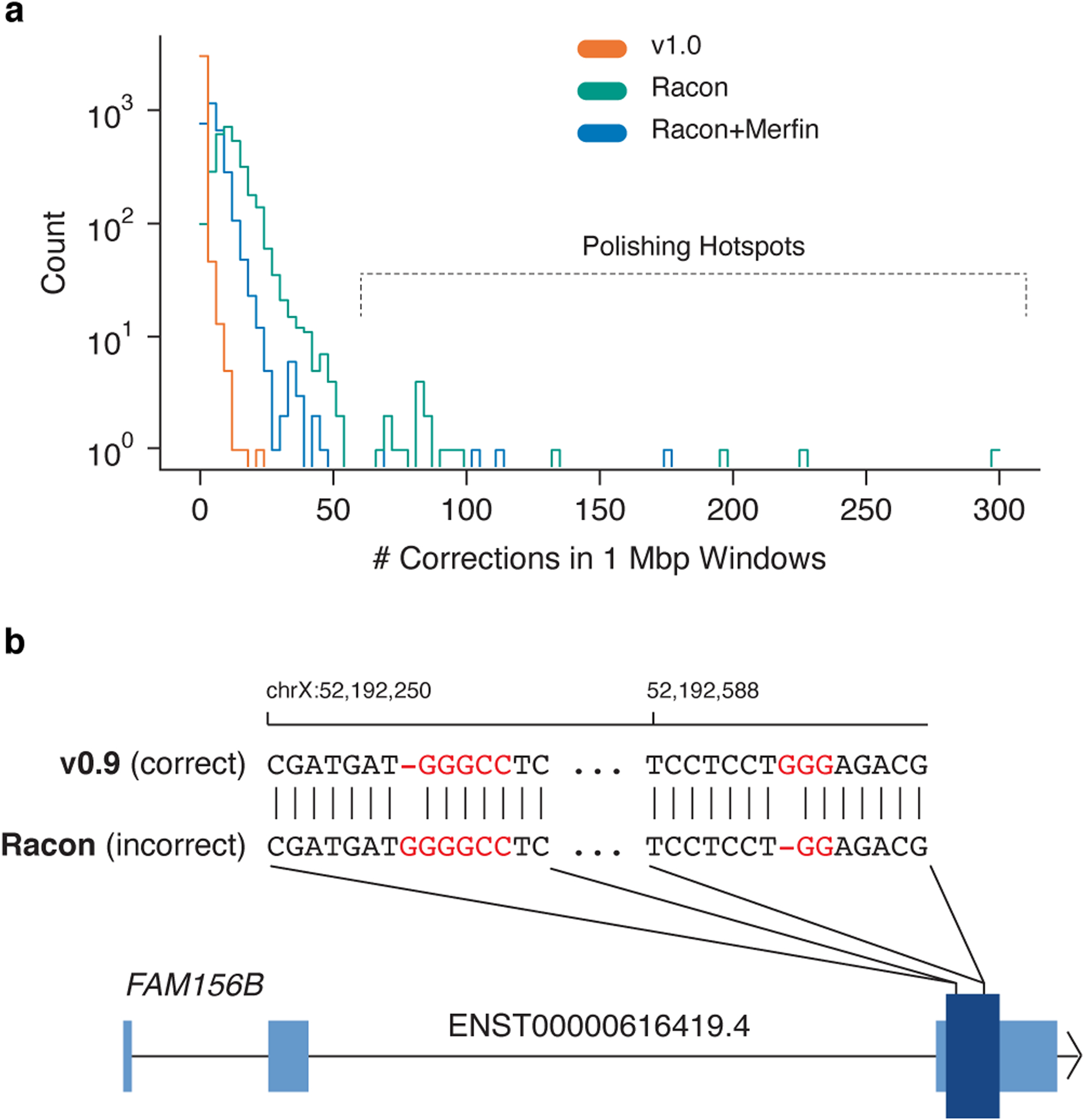
Advances in long-read sequencing technologies and genome assembly methods have enabled the recent completion of the first telomere-to-telomere human genome assembly, which resolves complex segmental duplications and large tandem repeats, including centromeric satellite arrays in a complete hydatidiform mole (CHM13). Although derived from highly accurate sequences, evaluation revealed evidence of small errors and structural misassemblies in the initial draft assembly. To correct these errors, we designed a new repeat-aware polishing strategy that made accurate assembly corrections in large repeats without overcorrection, ultimately fixing 51% of the existing errors and improving the assembly quality value from 70.2 to 73.9 measured from PacBio high-fidelity and Illumina k-mers. By comparing our results to standard automated polishing tools, we outline common polishing errors and offer practical suggestions for genome projects with limited resources. We also show how sequencing biases in both high-fidelity and Oxford Nanopore Technologies reads cause signature assembly errors that can be corrected with a diverse panel of sequencing technologies.
© 2022. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Figures
Comment in
-
Polishing high-quality genome assemblies.Nat Methods. 2022 Jun;19(6):649-650. doi: 10.1038/s41592-022-01515-1. Nat Methods. 2022. PMID: 35610477 No abstract available.
References
Publication types
MeSH terms
Grants and funding
- U01 HG010961/HG/NHGRI NIH HHS/United States
- WT206194/WT_/Wellcome Trust/United Kingdom
- U41 HG010972/HG/NHGRI NIH HHS/United States
- F32 GM134558/GM/NIGMS NIH HHS/United States
- OT2 OD026682/OD/NIH HHS/United States
- U24 HG007234/HG/NHGRI NIH HHS/United States
- R01 HG011274/HG/NHGRI NIH HHS/United States
- R21 HG010548/HG/NHGRI NIH HHS/United States
- U01 HG010971/HG/NHGRI NIH HHS/United States
- U24 HG011853/HG/NHGRI NIH HHS/United States
- R01 HG010485/HG/NHGRI NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- ZIA HG200398/ImNIH/Intramural NIH HHS/United States
- R01 HG006677/HG/NHGRI NIH HHS/United States
- Z99 HG999999/ImNIH/Intramural NIH HHS/United States
LinkOut - more resources
Full Text Sources
