

Merfin: improved variant filtering, assembly evaluation and polishing via k-mer validation
- PMID: 35361932
- PMCID: PMC9745813
- DOI: 10.1038/s41592-022-01445-y
Merfin: improved variant filtering, assembly evaluation and polishing via k-mer validation
Abstract
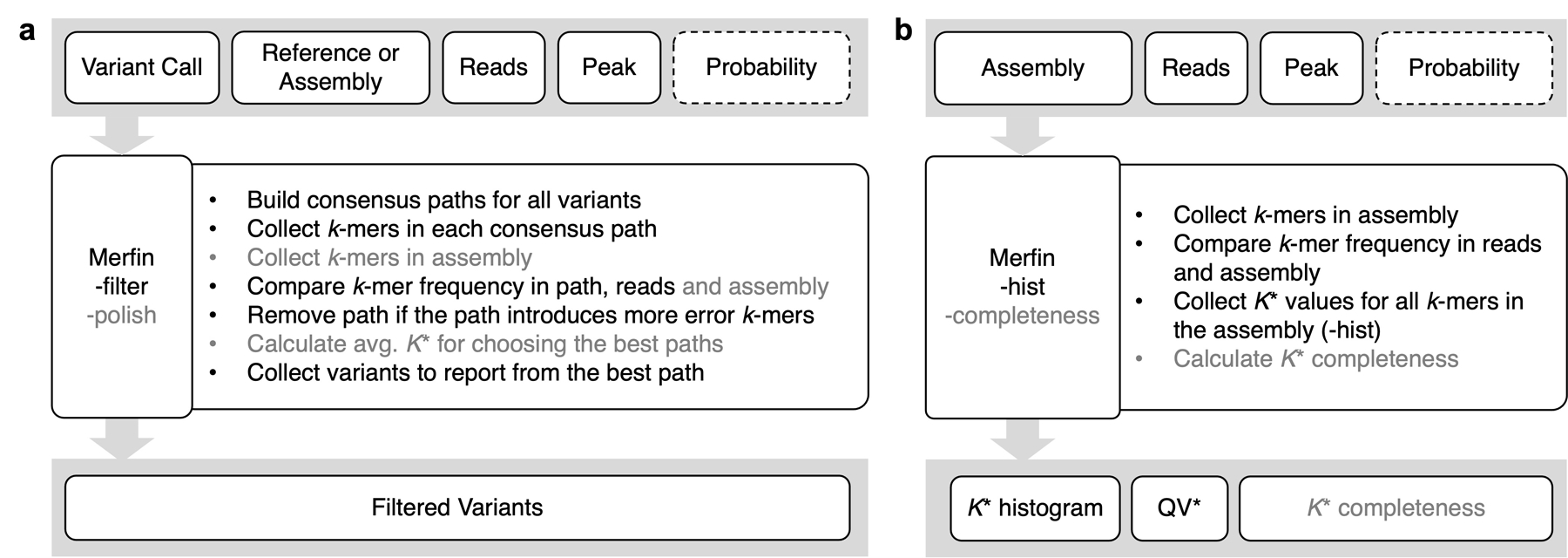
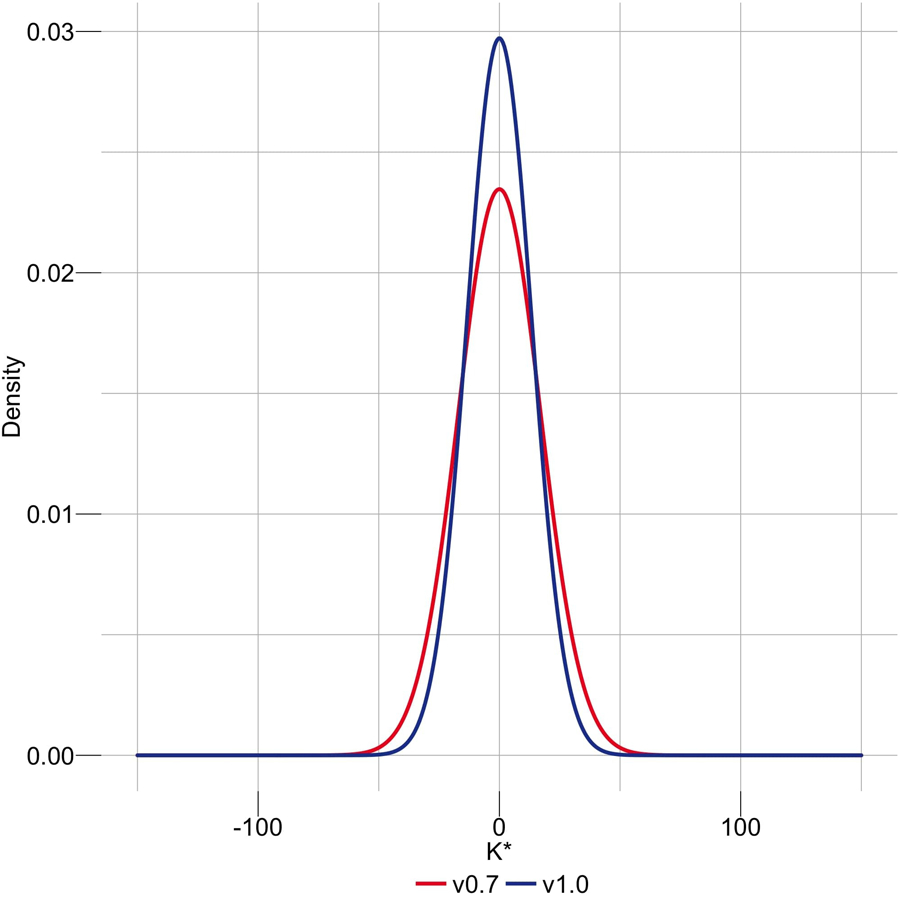
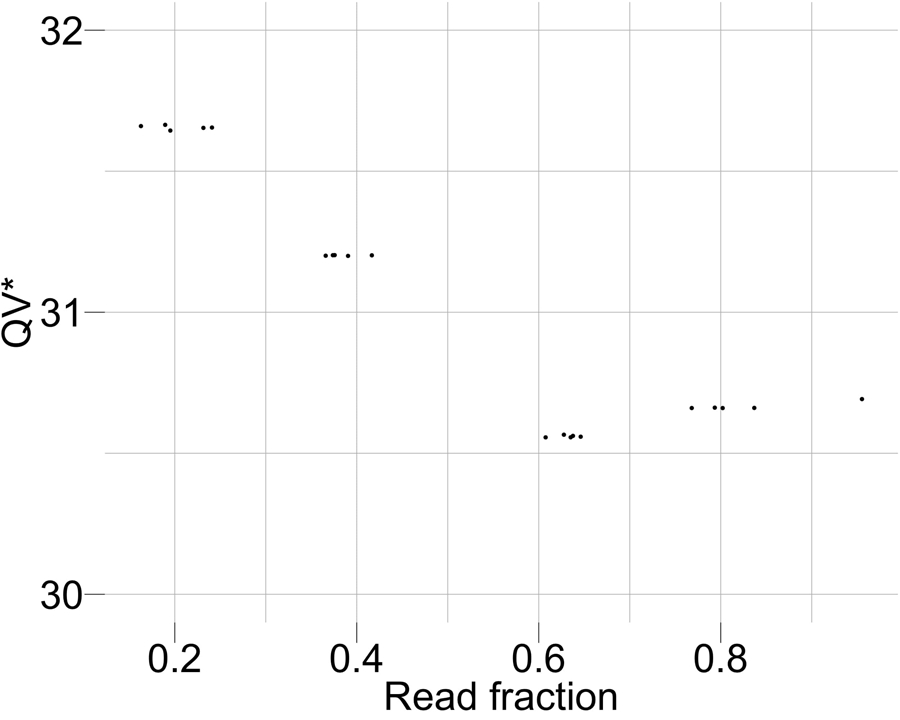
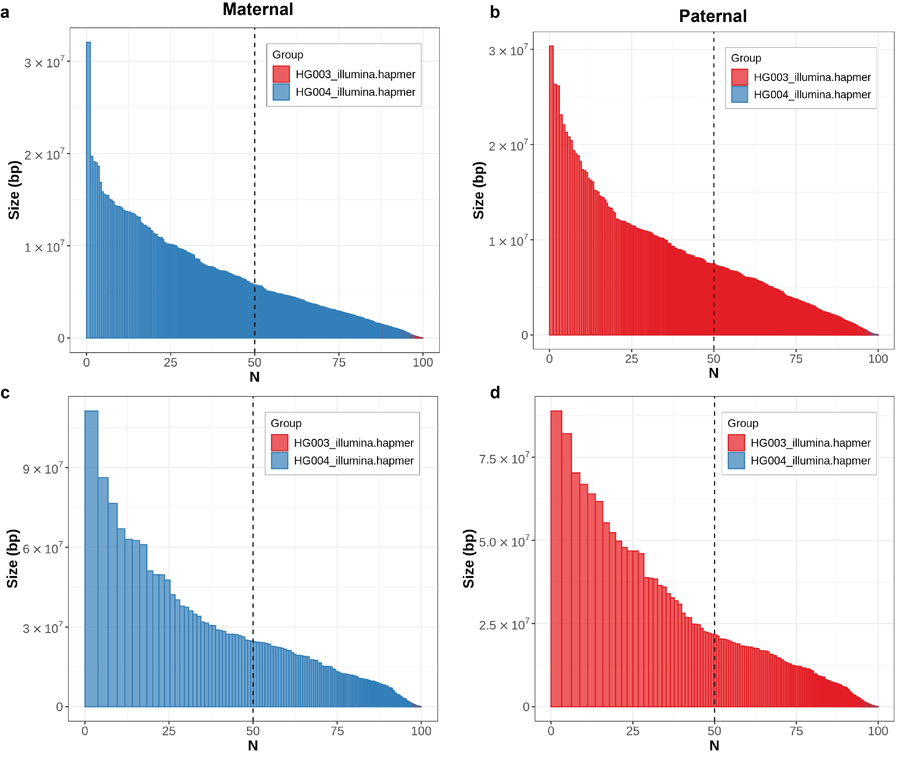
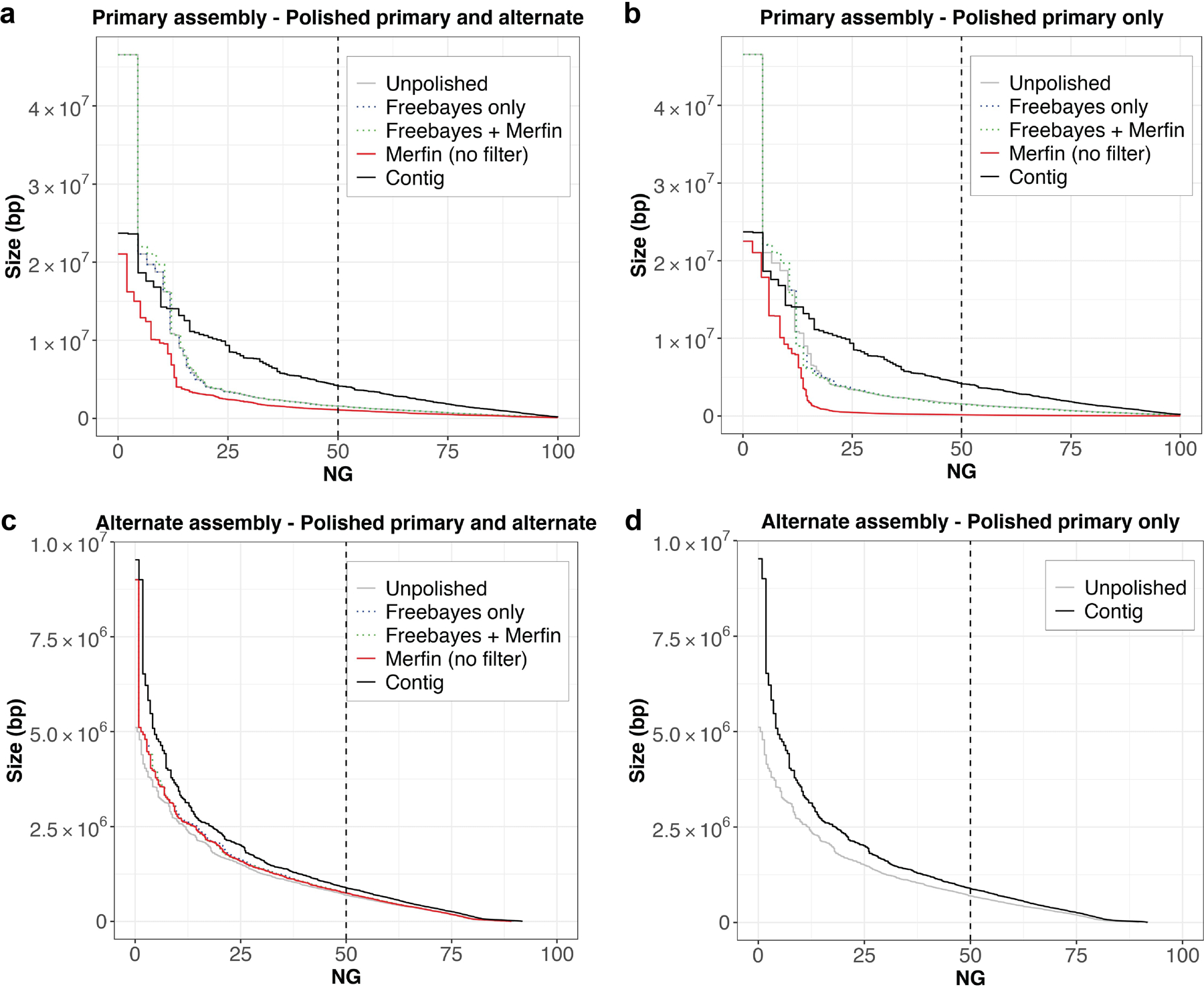
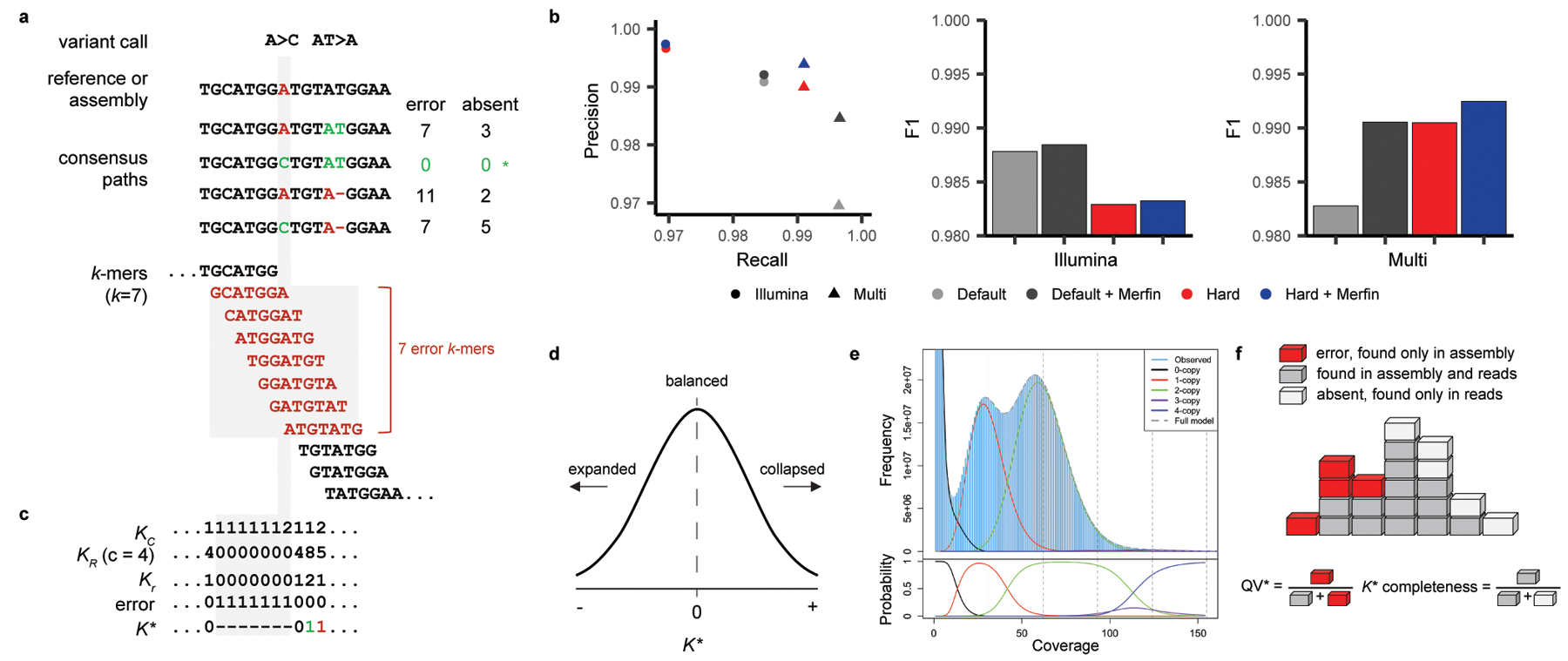
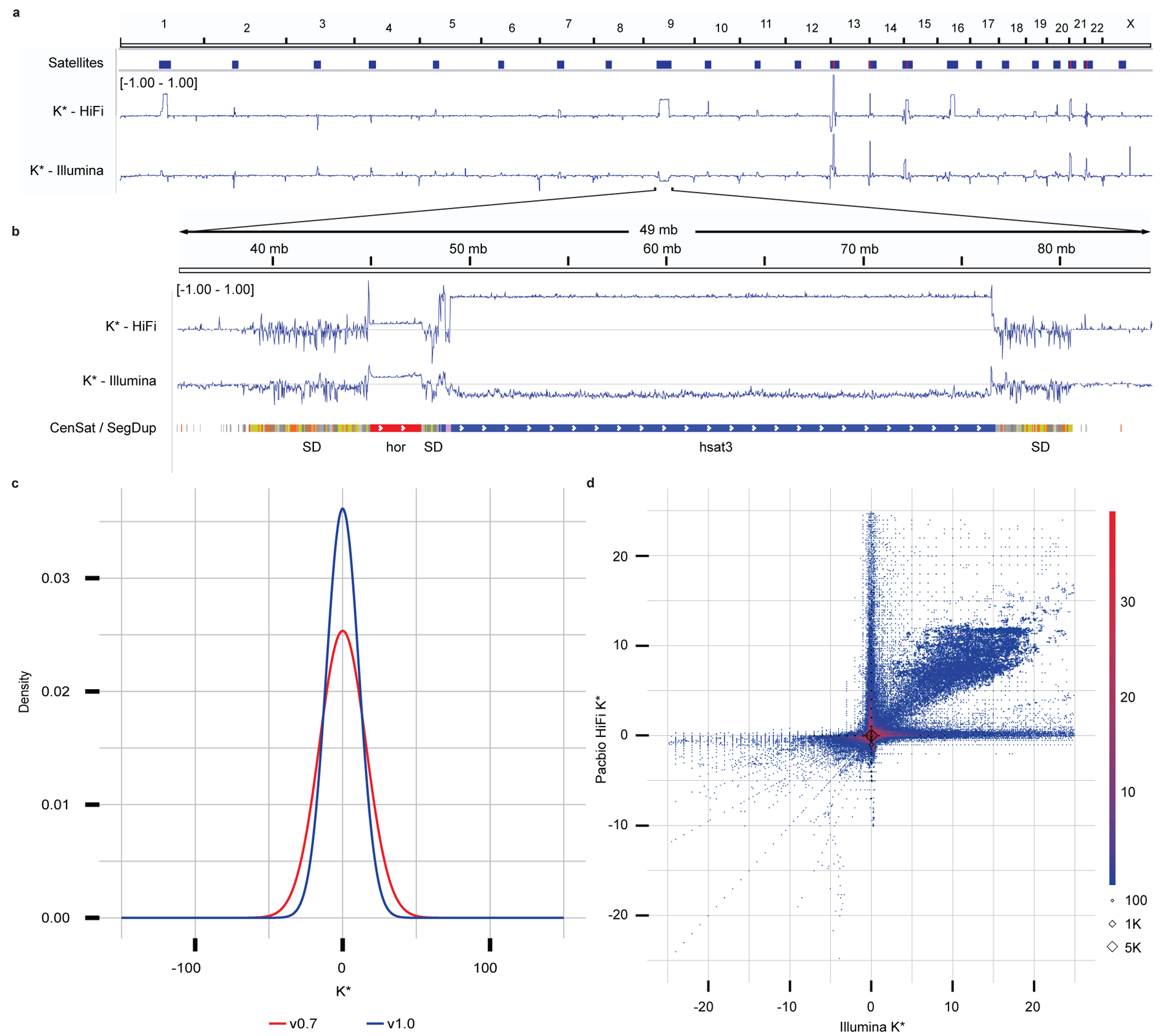
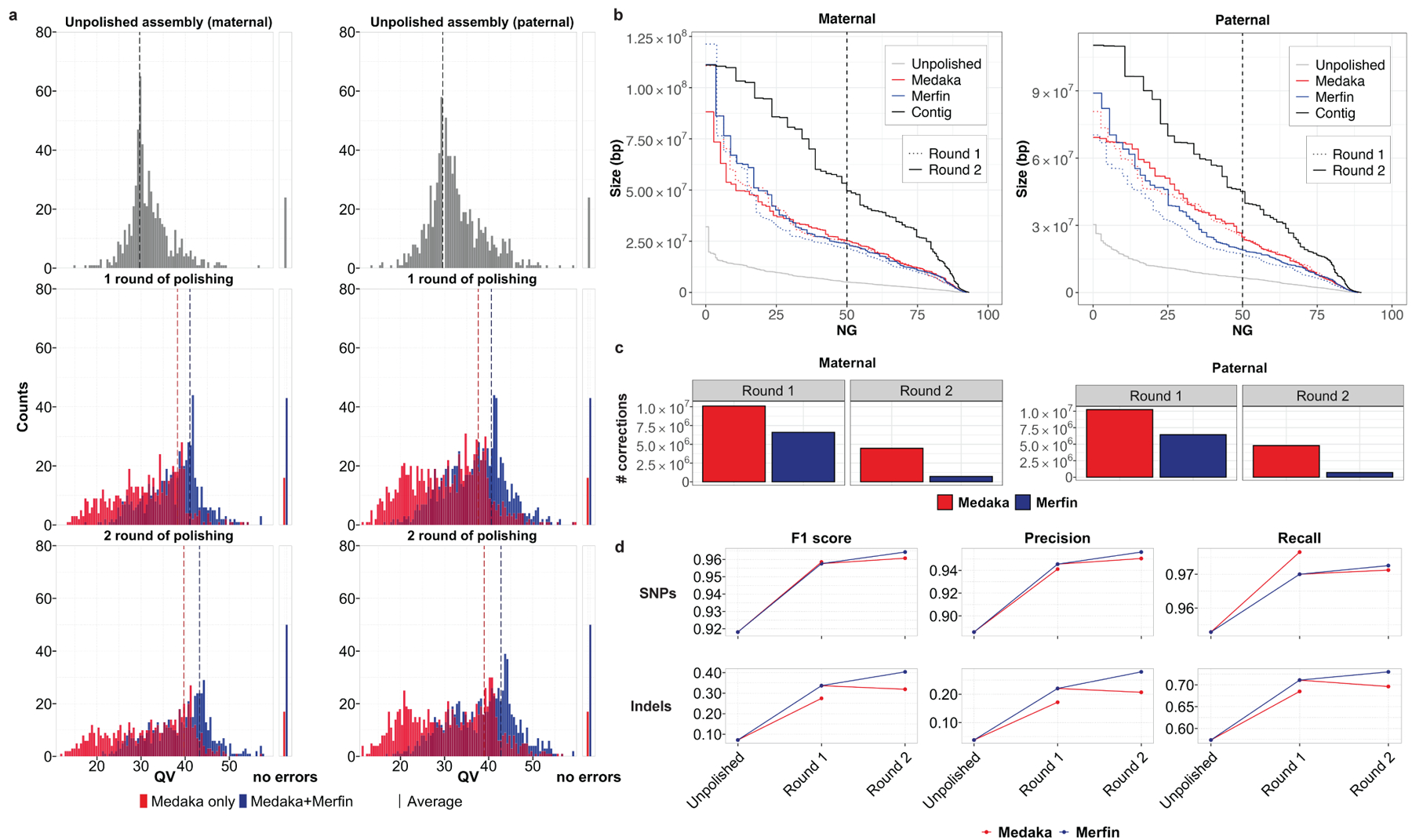
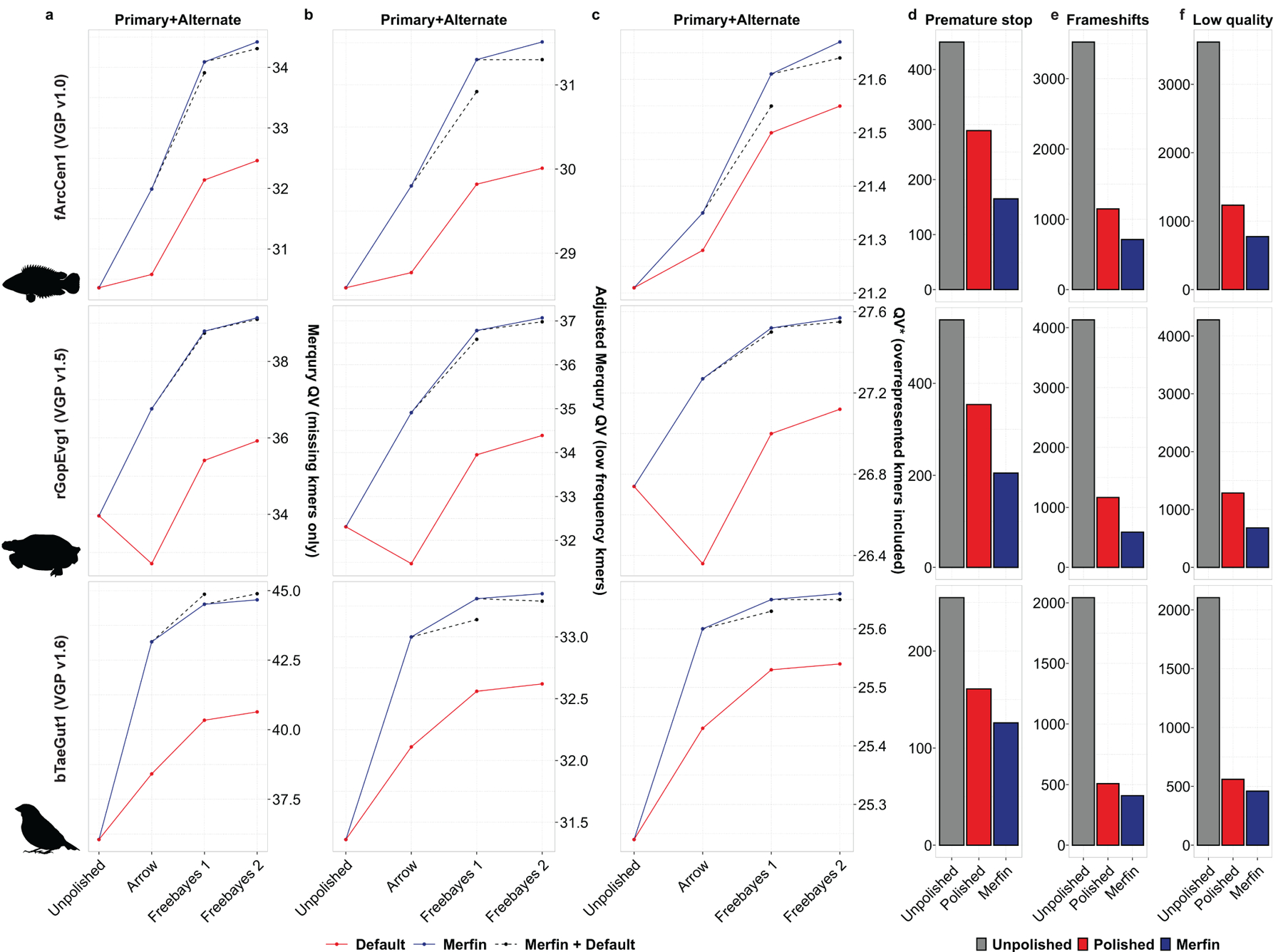
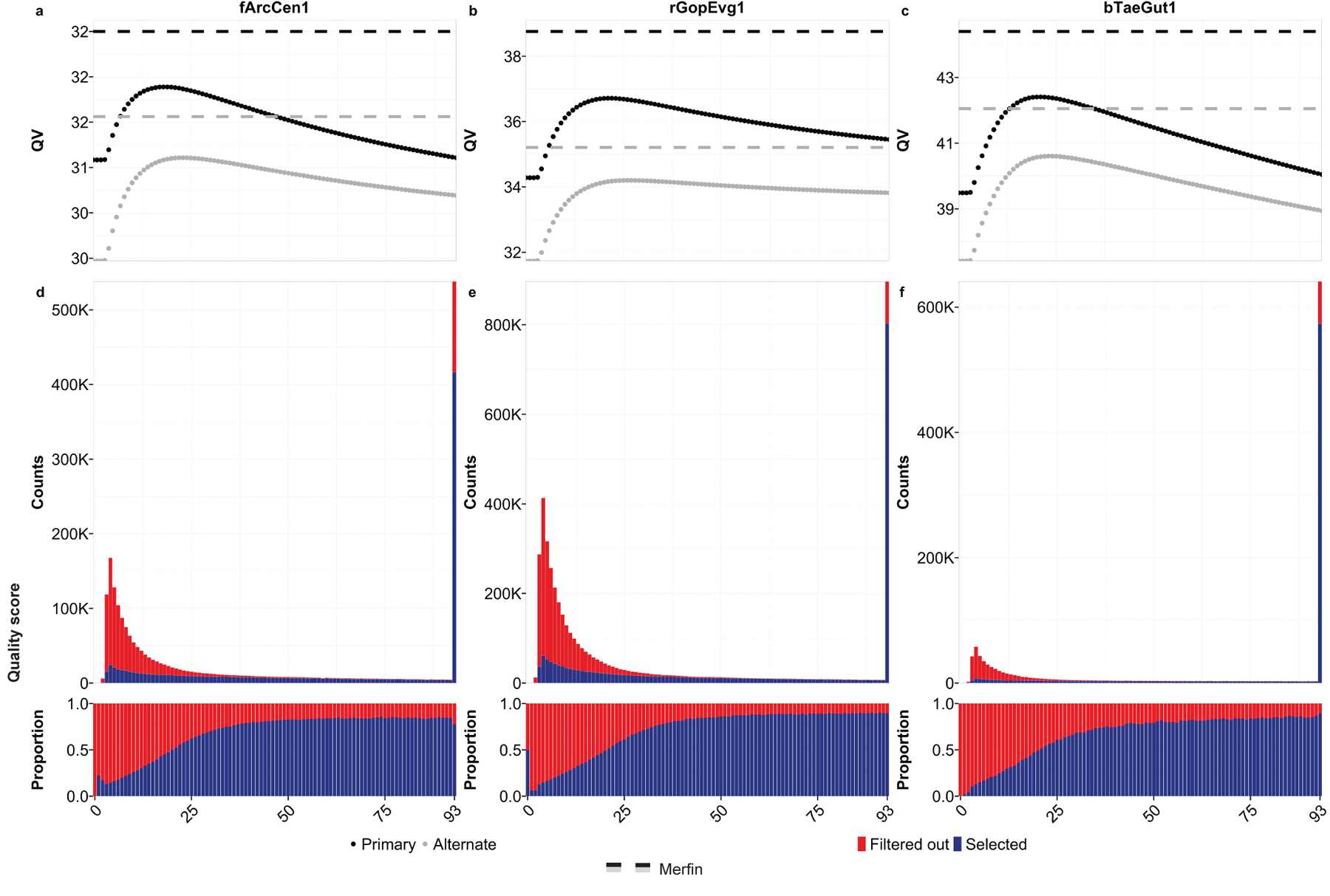
Variant calling has been widely used for genotyping and for improving the consensus accuracy of long-read assemblies. Variant calls are commonly hard-filtered with user-defined cutoffs. However, it is impossible to define a single set of optimal cutoffs, as the calls heavily depend on the quality of the reads, the variant caller of choice and the quality of the unpolished assembly. Here, we introduce Merfin, a k-mer based variant-filtering algorithm for improved accuracy in genotyping and genome assembly polishing. Merfin evaluates each variant based on the expected k-mer multiplicity in the reads, independently of the quality of the read alignment and variant caller's internal score. Merfin increased the precision of genotyped calls in several benchmarks, improved consensus accuracy and reduced frameshift errors when applied to human and nonhuman assemblies built from Pacific Biosciences HiFi and continuous long reads or Oxford Nanopore reads, including the first complete human genome. Moreover, we introduce assembly quality and completeness metrics that account for the expected genomic copy numbers.
© 2022. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
Competing Interests Statement
S.K. has received travel funds to speak at symposia organized by Oxford Nanopore. The remaining authors declare no competing interests.
Figures



References
Methods References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
