

Polygenic prediction of educational attainment within and between families from genome-wide association analyses in 3 million individuals
- PMID: 35361970
- PMCID: PMC9005349
- DOI: 10.1038/s41588-022-01016-z
Polygenic prediction of educational attainment within and between families from genome-wide association analyses in 3 million individuals
Abstract
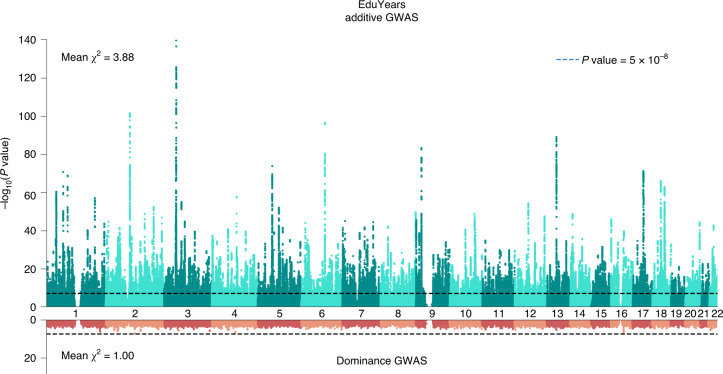
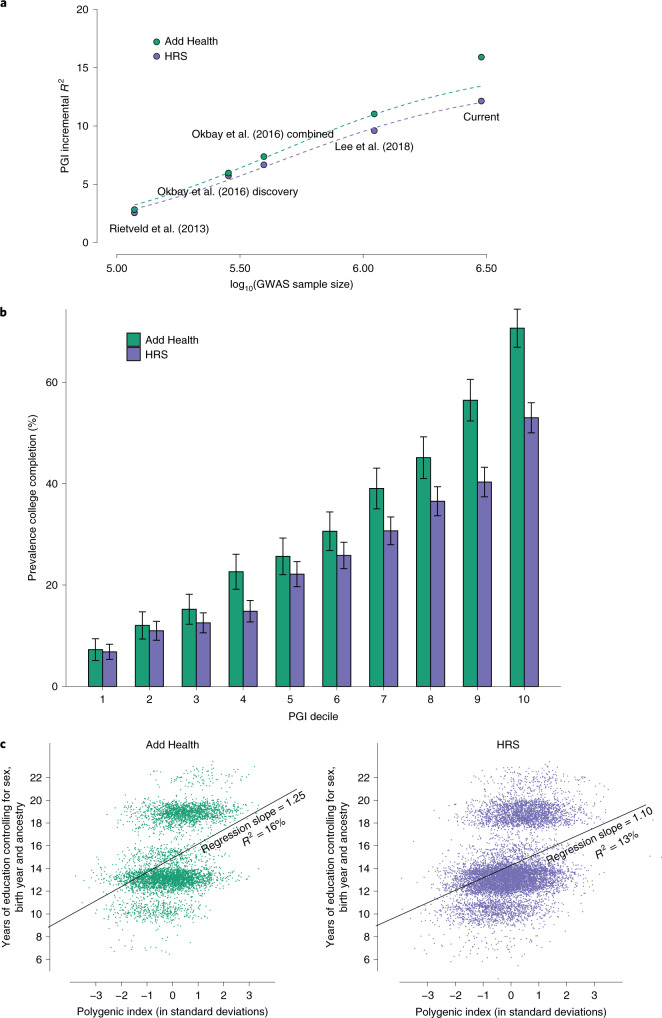
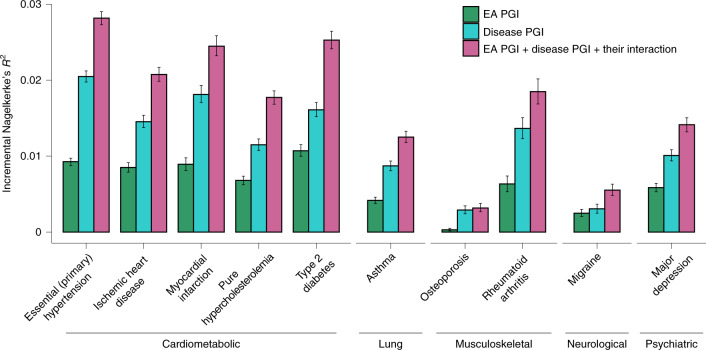
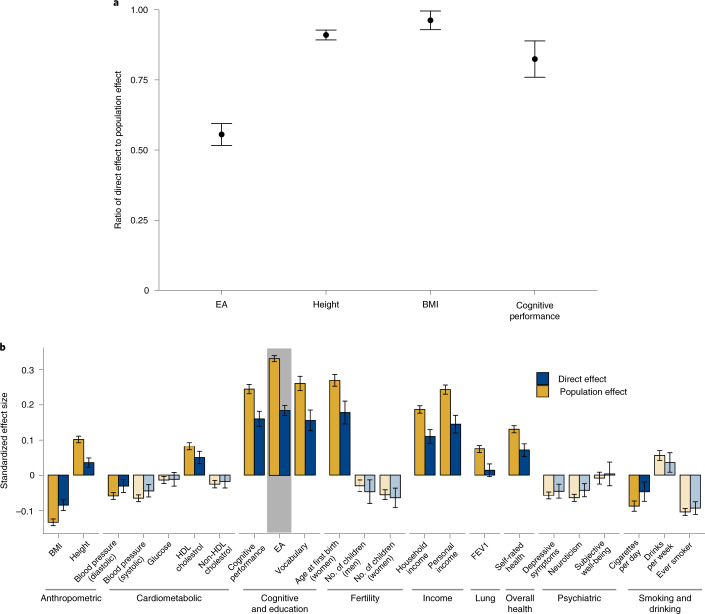
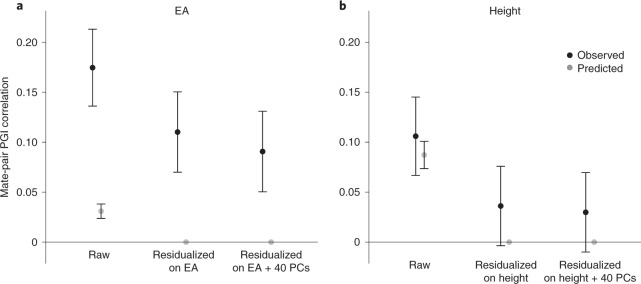
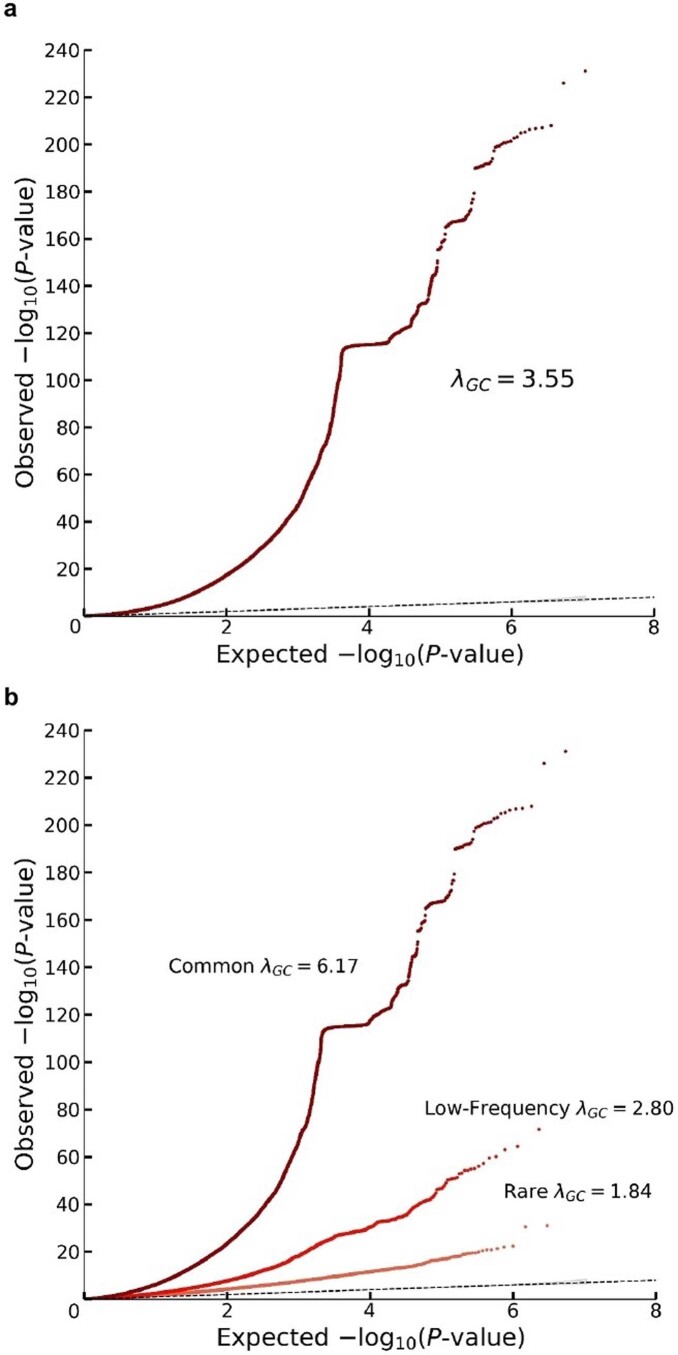
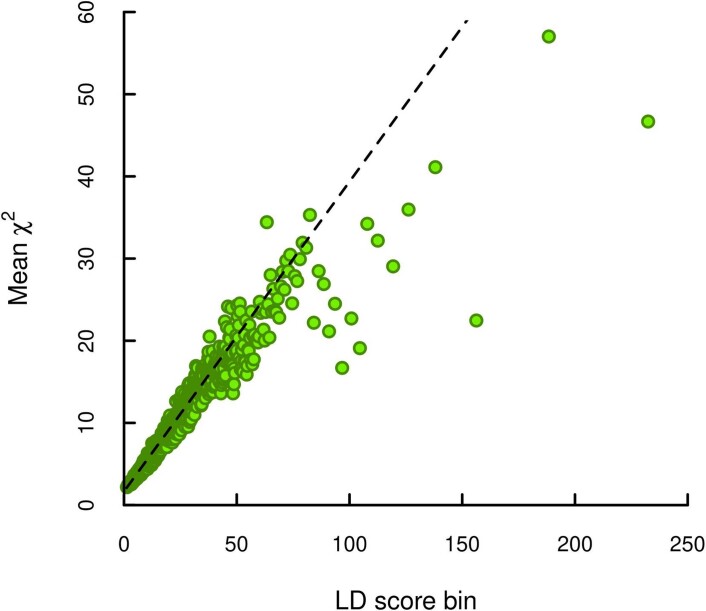
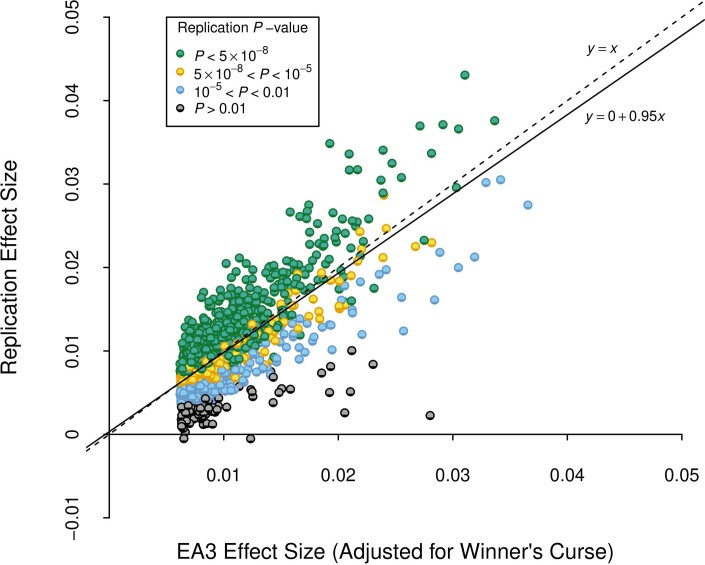
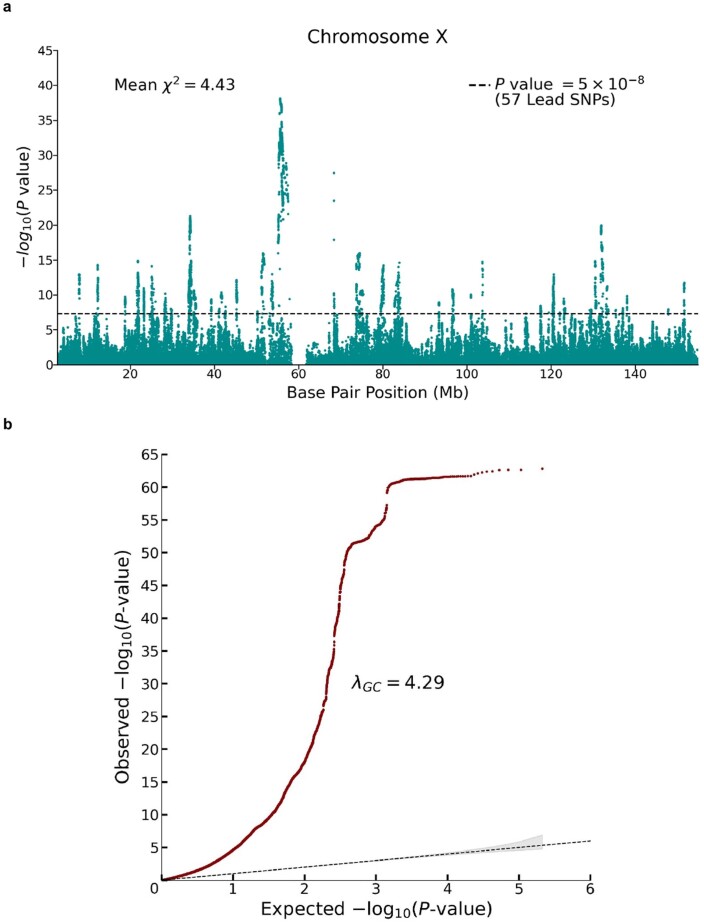
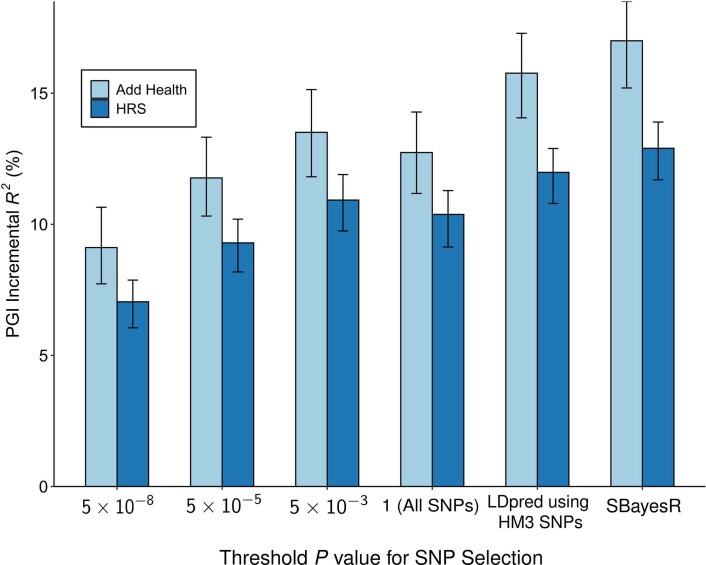
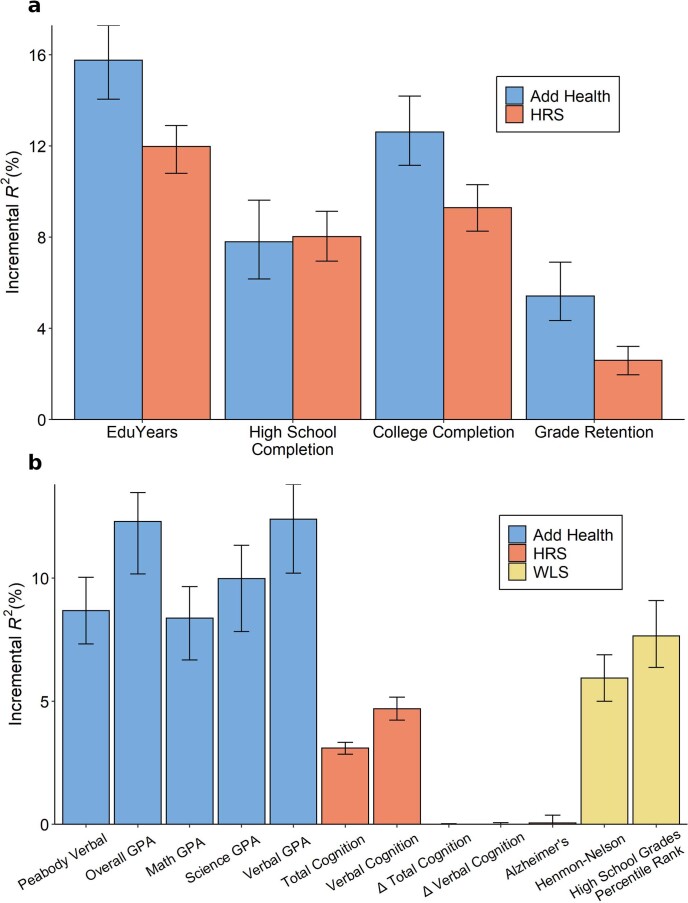
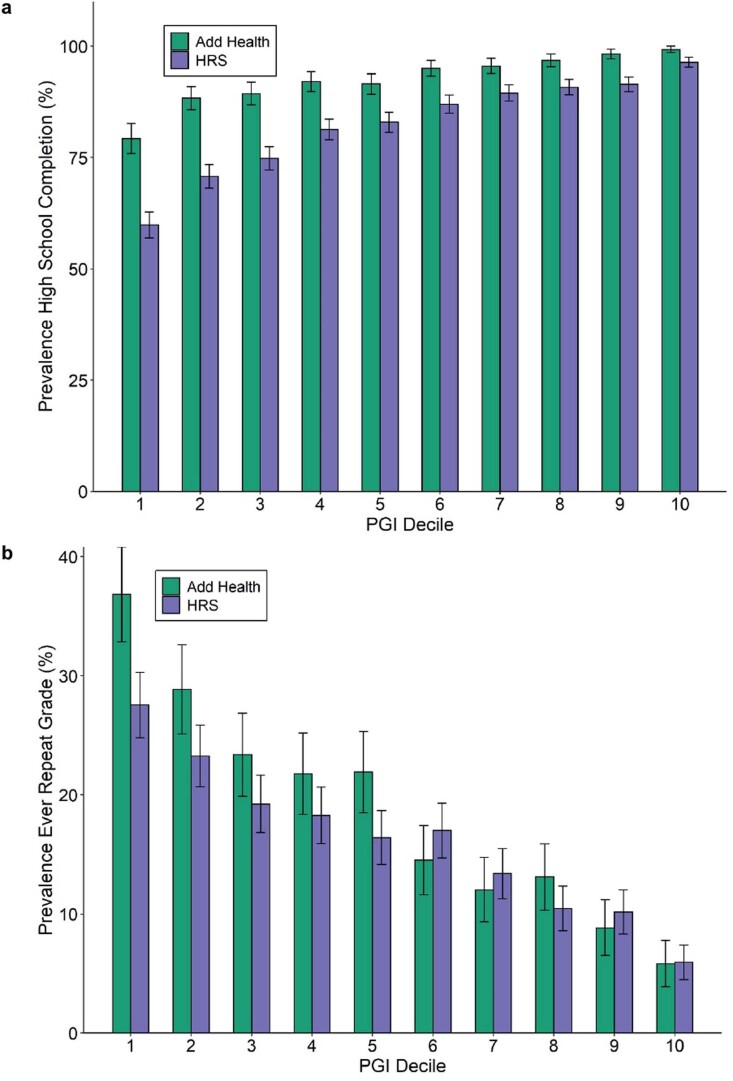
We conduct a genome-wide association study (GWAS) of educational attainment (EA) in a sample of ~3 million individuals and identify 3,952 approximately uncorrelated genome-wide-significant single-nucleotide polymorphisms (SNPs). A genome-wide polygenic predictor, or polygenic index (PGI), explains 12-16% of EA variance and contributes to risk prediction for ten diseases. Direct effects (i.e., controlling for parental PGIs) explain roughly half the PGI's magnitude of association with EA and other phenotypes. The correlation between mate-pair PGIs is far too large to be consistent with phenotypic assortment alone, implying additional assortment on PGI-associated factors. In an additional GWAS of dominance deviations from the additive model, we identify no genome-wide-significant SNPs, and a separate X-chromosome additive GWAS identifies 57.
© 2022. The Author(s).
Conflict of interest statement
Y.J., B.H., C.T., D.A.H. and the members of the 23andMe Research Team are current or former employees of 23andMe, Inc. All other authors declare no competing interests.
Figures
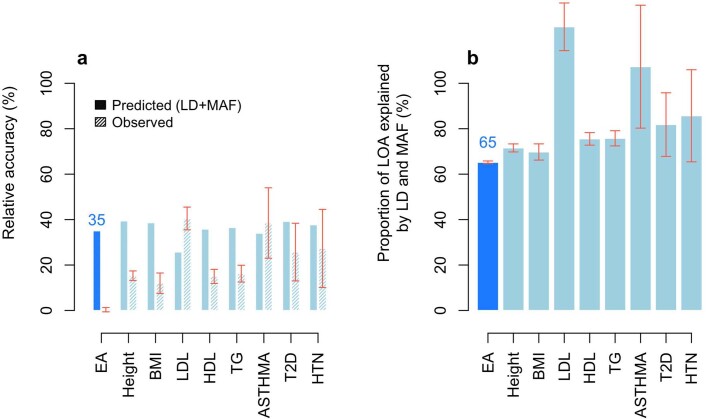
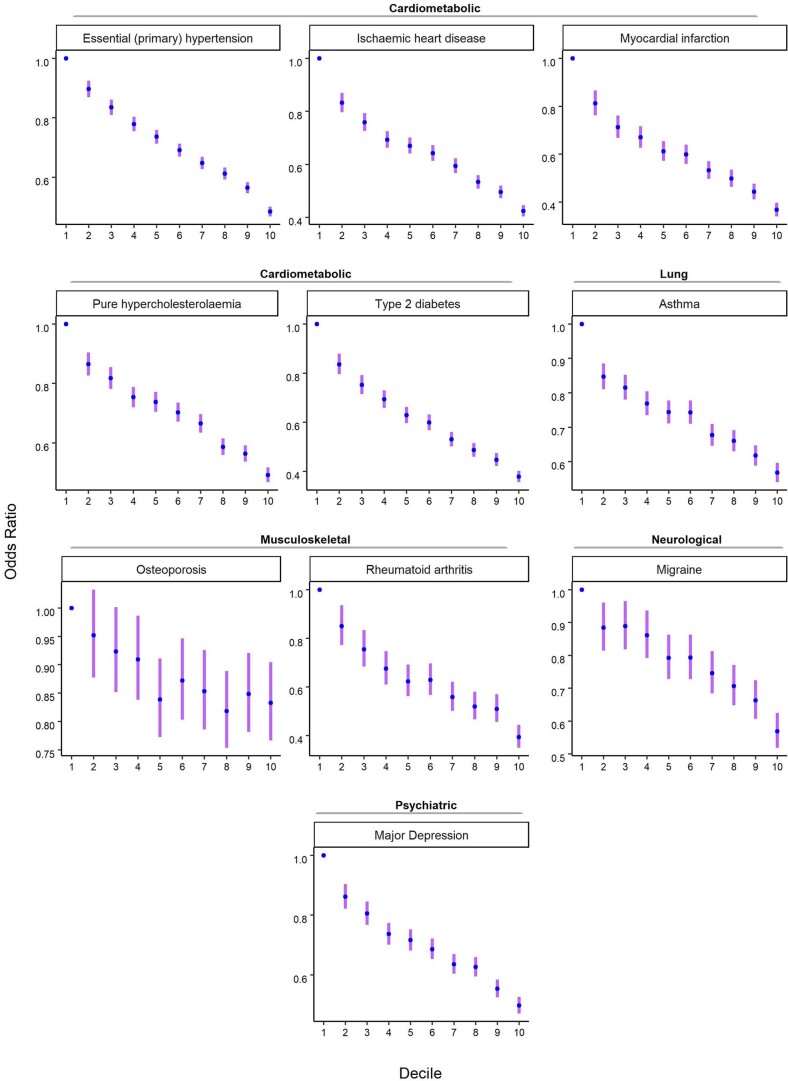
Comment in
-
Indirect paths from genetics to education.Nat Genet. 2022 Apr;54(4):372-373. doi: 10.1038/s41588-021-00999-5. Nat Genet. 2022. PMID: 35361971 No abstract available.
References
Publication types
MeSH terms
Grants and funding
- P30 AG072975/AG/NIA NIH HHS/United States
- K99 AG062787/AG/NIA NIH HHS/United States
- R24 AG065184/AG/NIA NIH HHS/United States
- R56 AG042568/AG/NIA NIH HHS/United States
- P30 AG010161/AG/NIA NIH HHS/United States
- MC_UU_00007/10/MRC_/Medical Research Council/United Kingdom
- R01 AG034374/AG/NIA NIH HHS/United States
- U01 MH109539/MH/NIMH NIH HHS/United States
- R01 MH101244/MH/NIMH NIH HHS/United States
- R00 AG062787/AG/NIA NIH HHS/United States
- R01 AG017917/AG/NIA NIH HHS/United States
- R01 AG042568/AG/NIA NIH HHS/United States
- MR/S019669/1/MRC_/Medical Research Council/United Kingdom
- R56 AG058726/AG/NIA NIH HHS/United States
- R01 AG015819/AG/NIA NIH HHS/United States
