

Inferring gene expression from cell-free DNA fragmentation profiles
- PMID: 35361996
- PMCID: PMC9337986
- DOI: 10.1038/s41587-022-01222-4
Inferring gene expression from cell-free DNA fragmentation profiles
Abstract
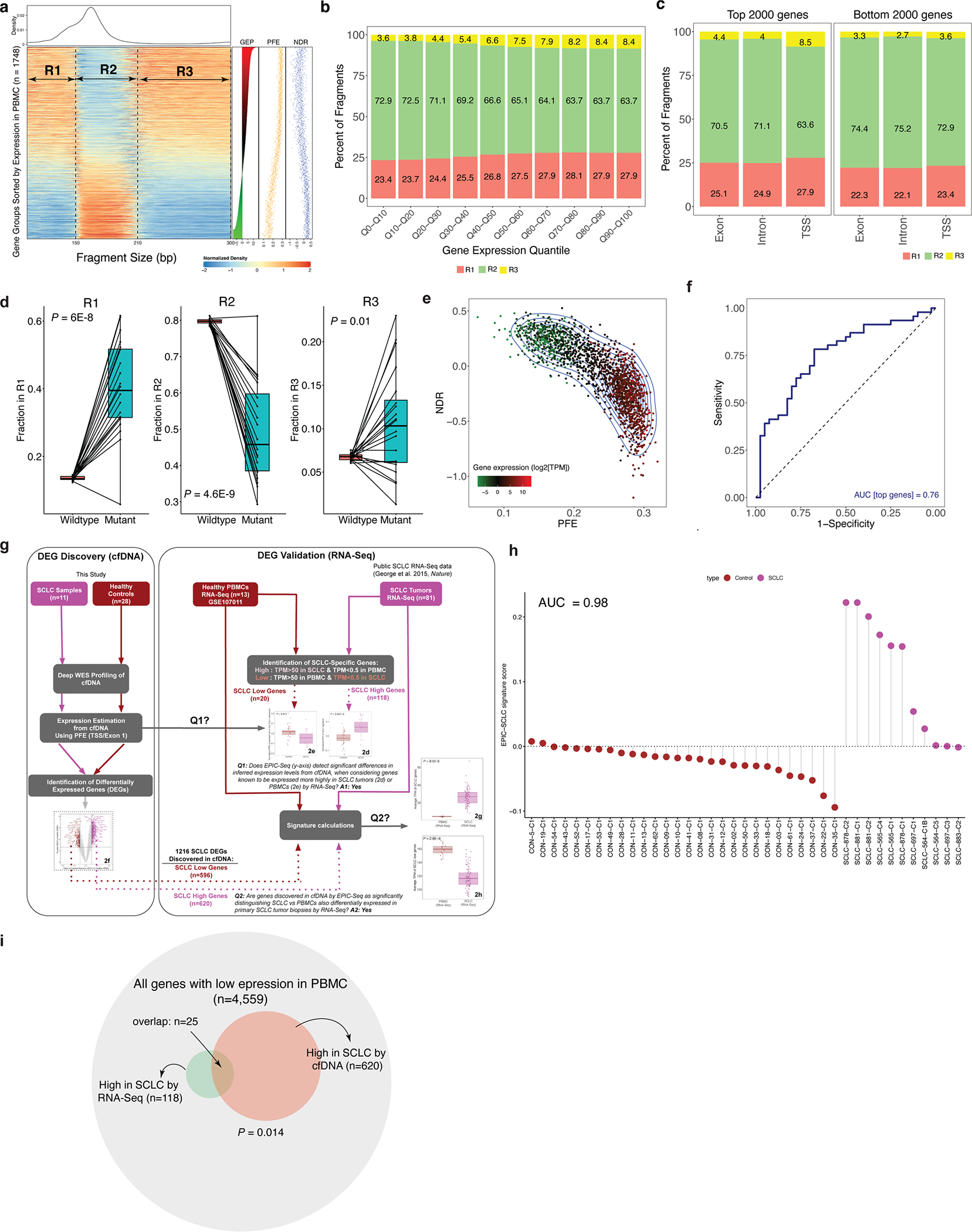
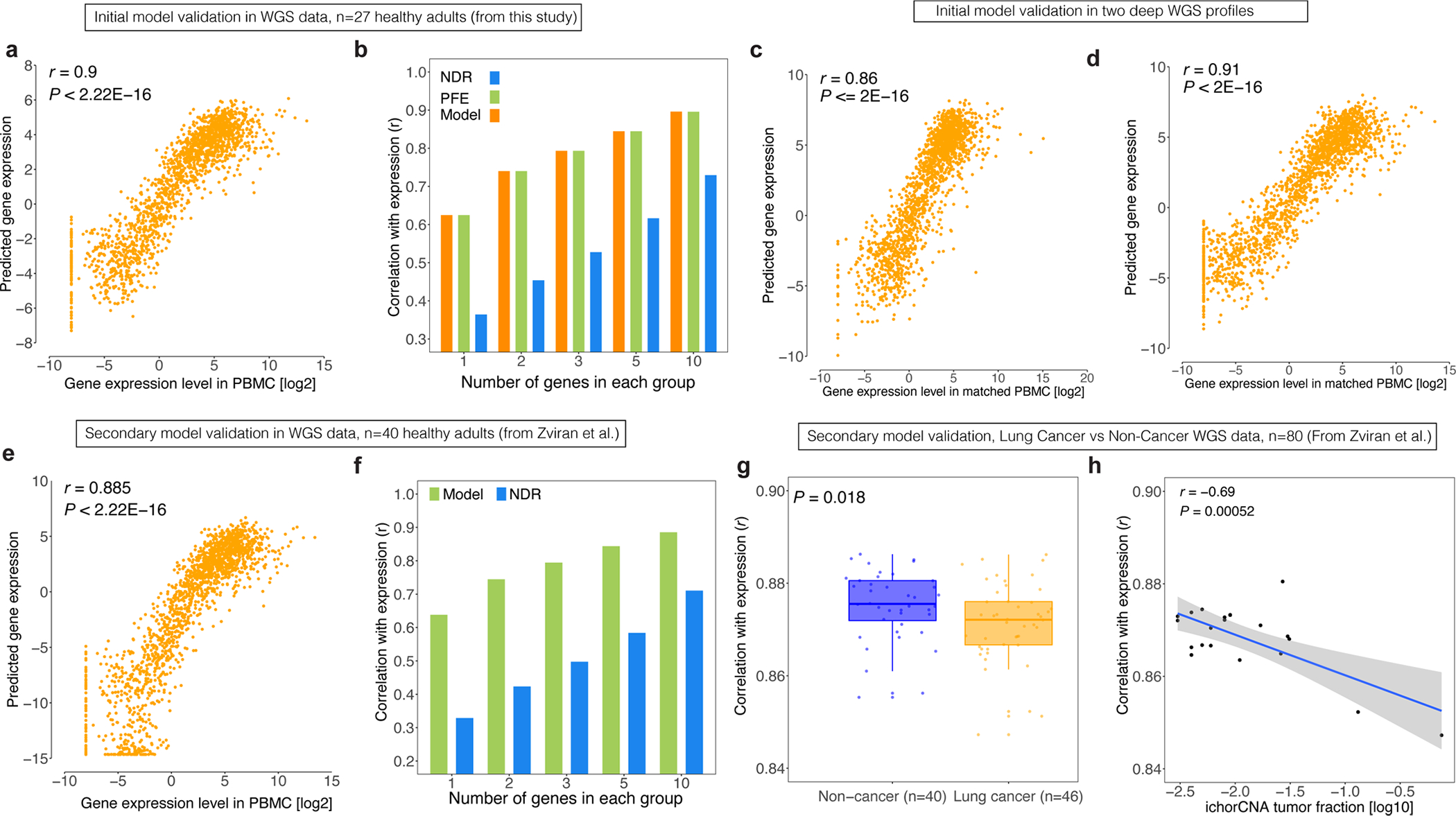
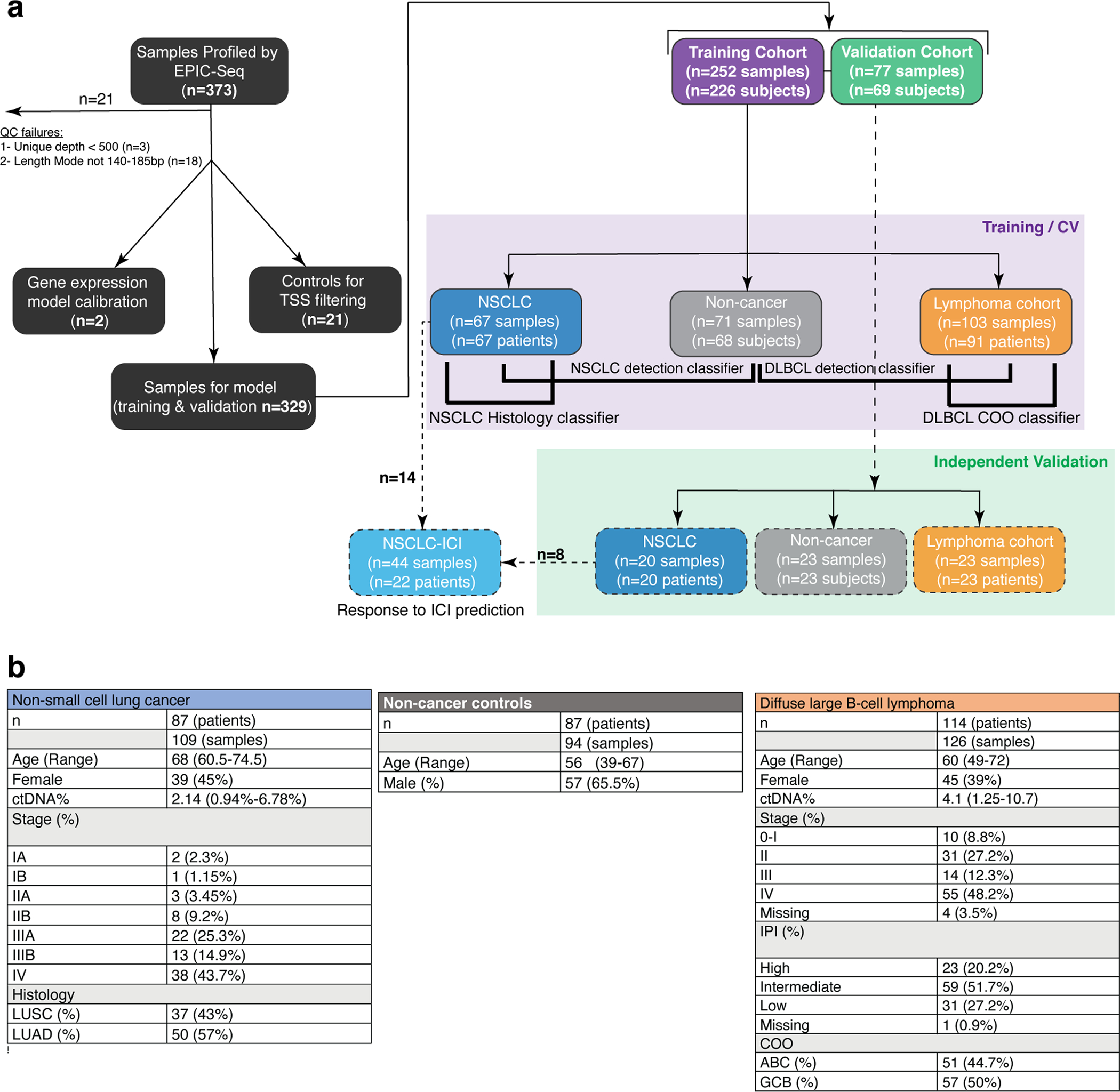
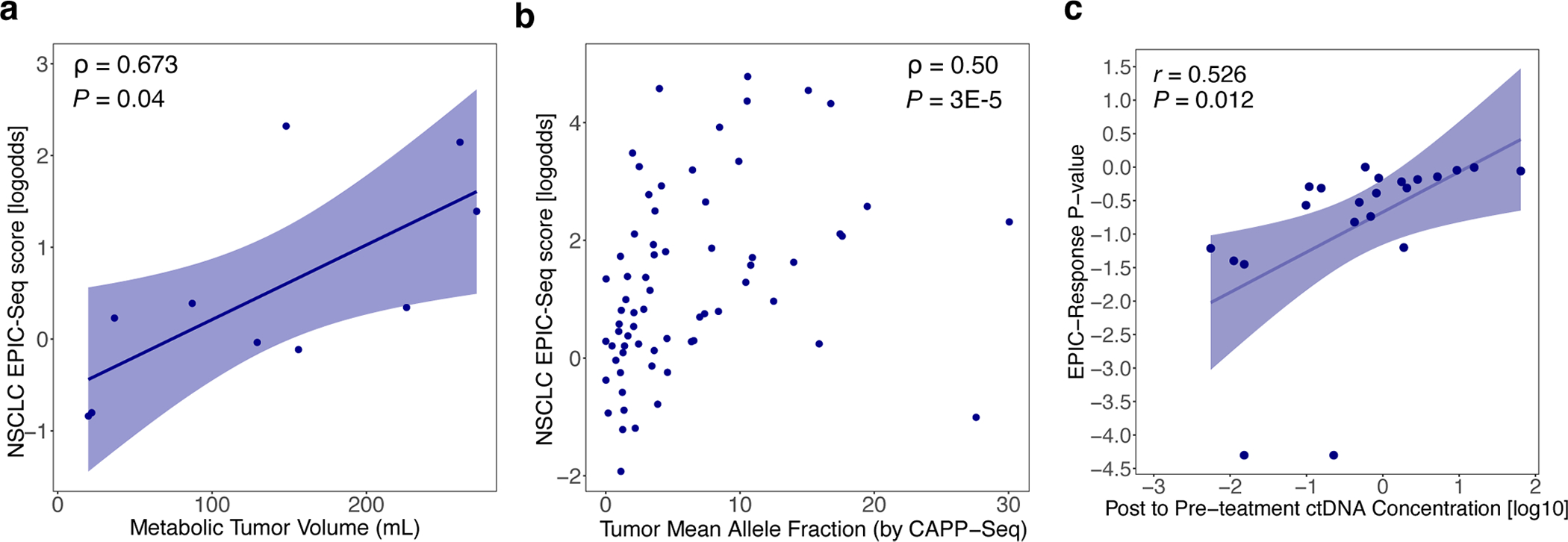
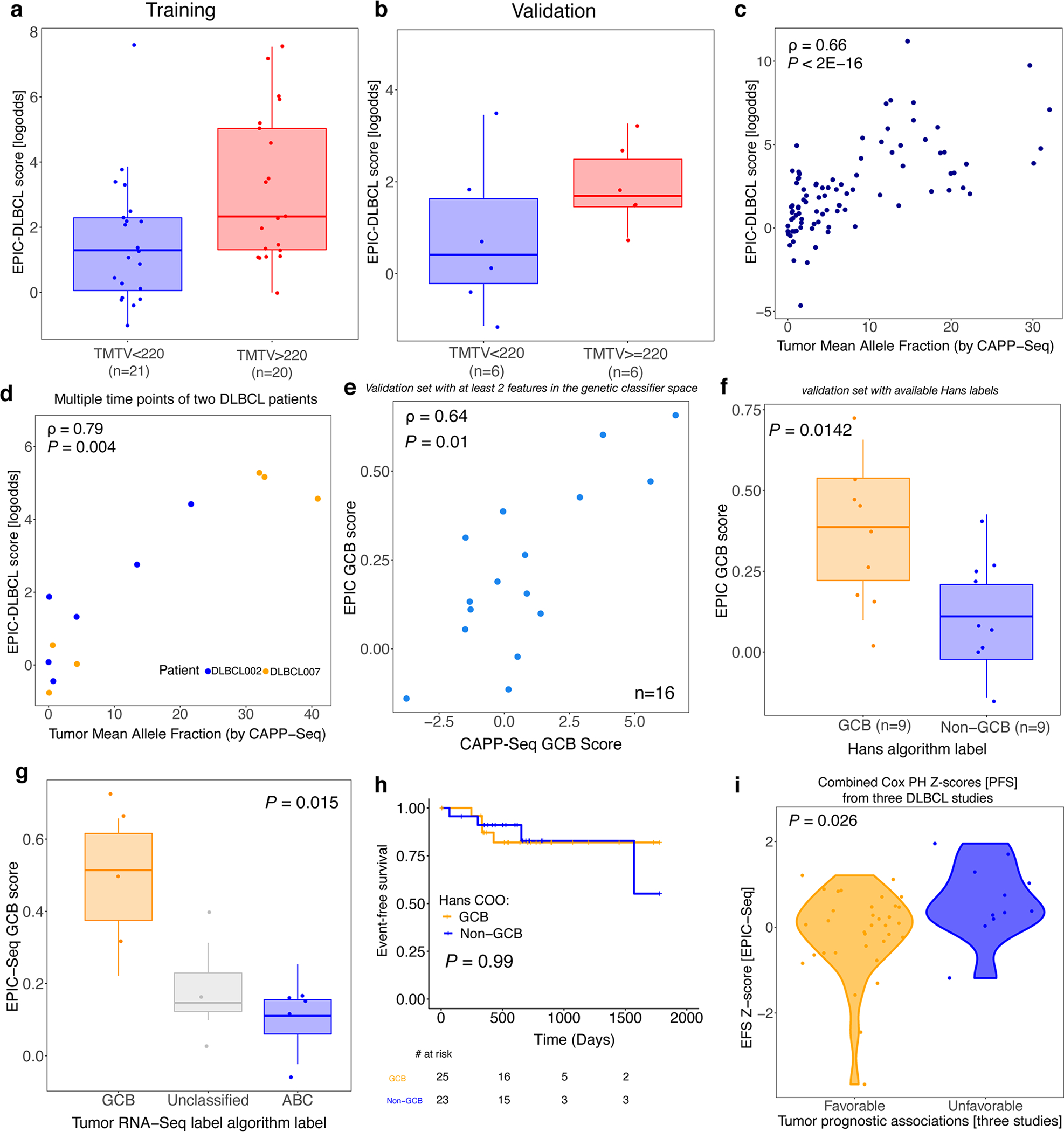
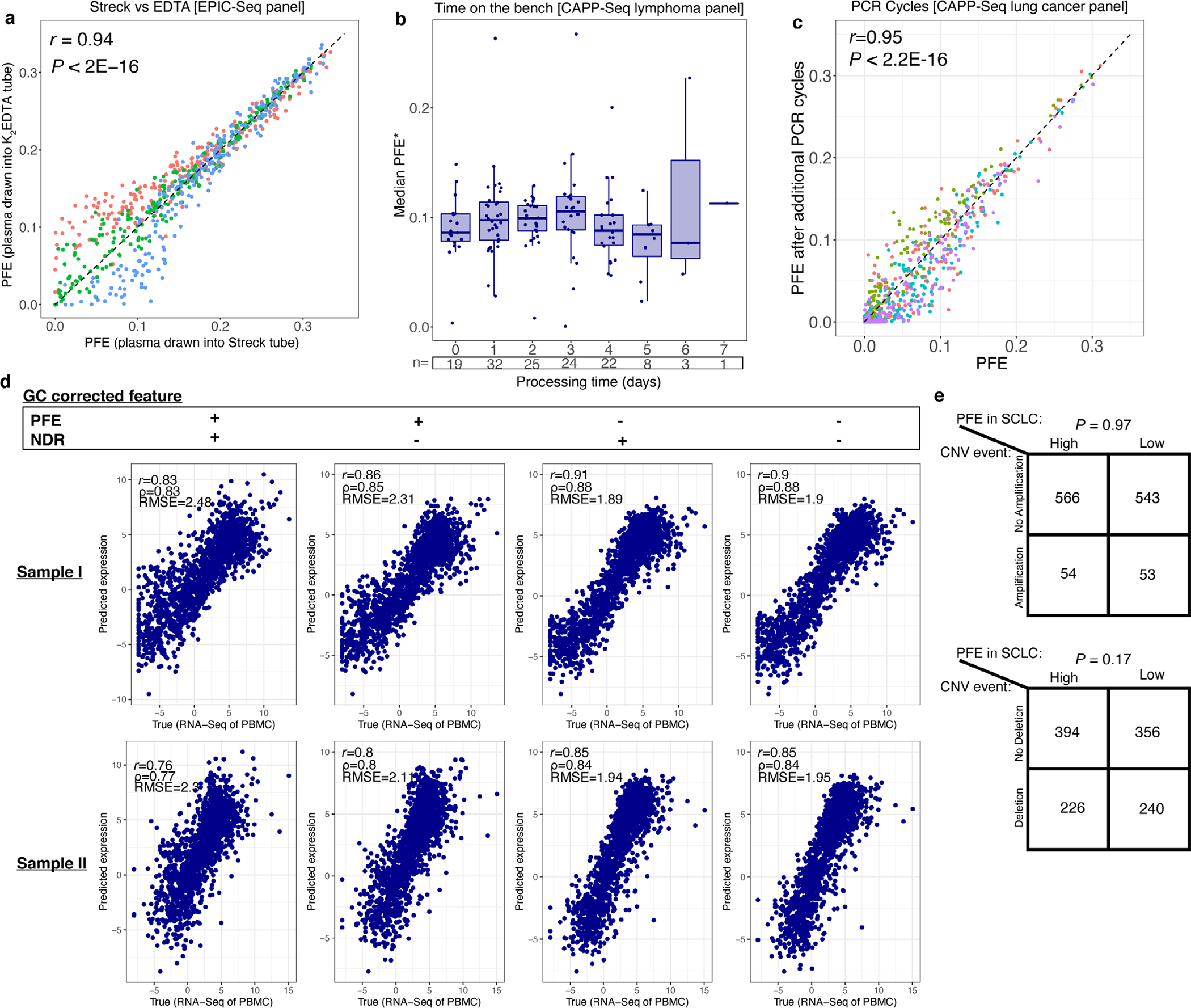
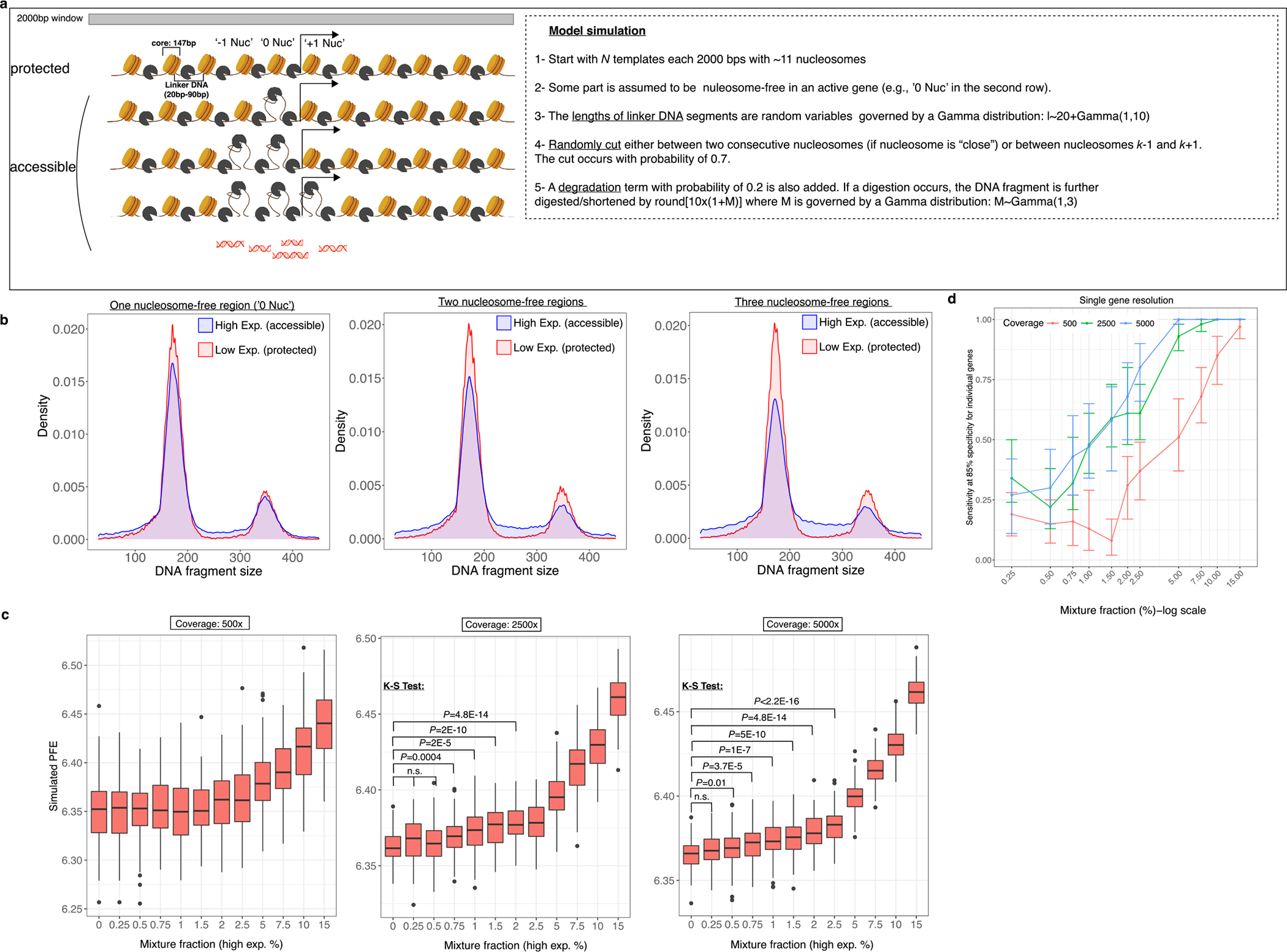
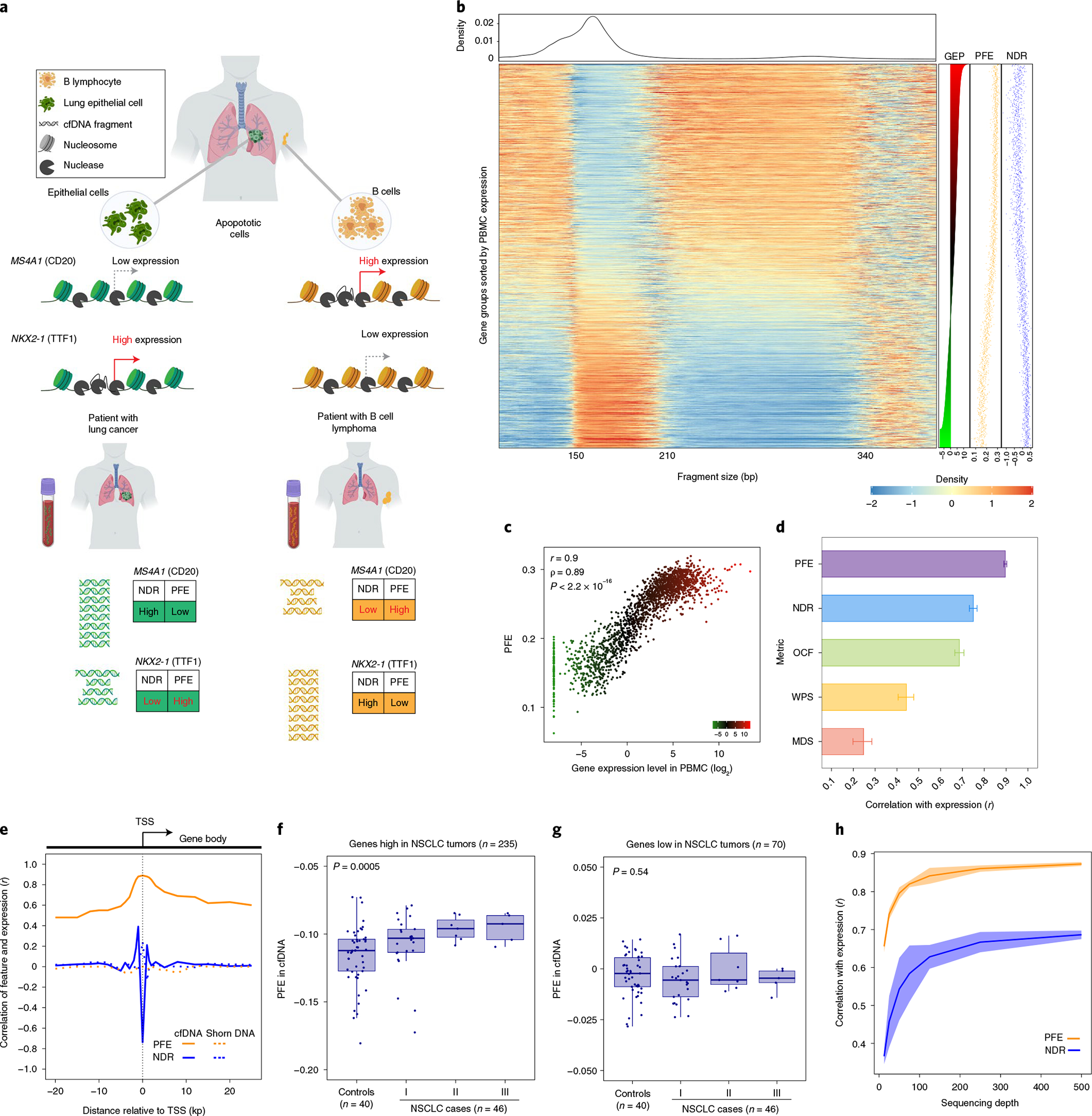
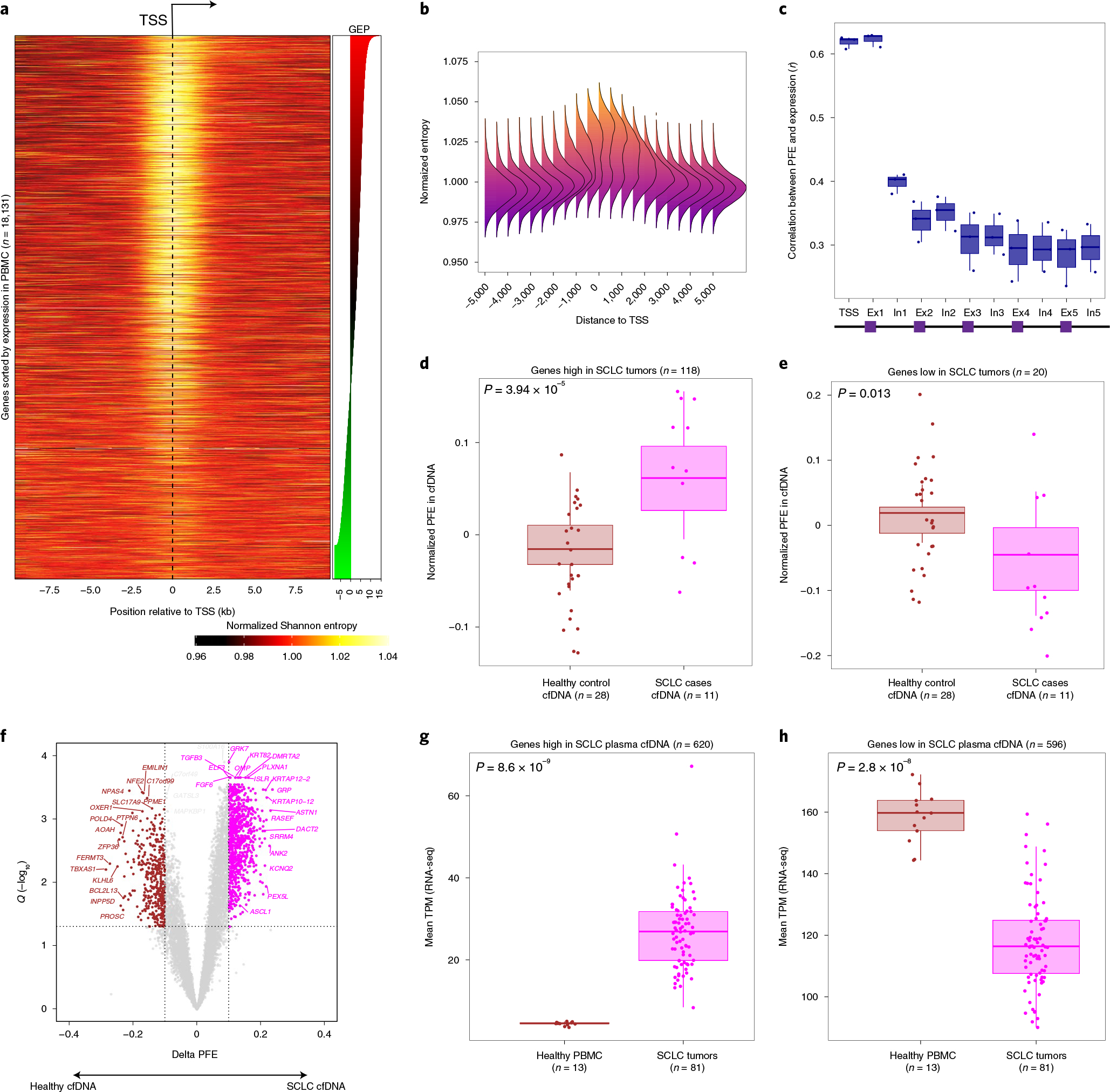
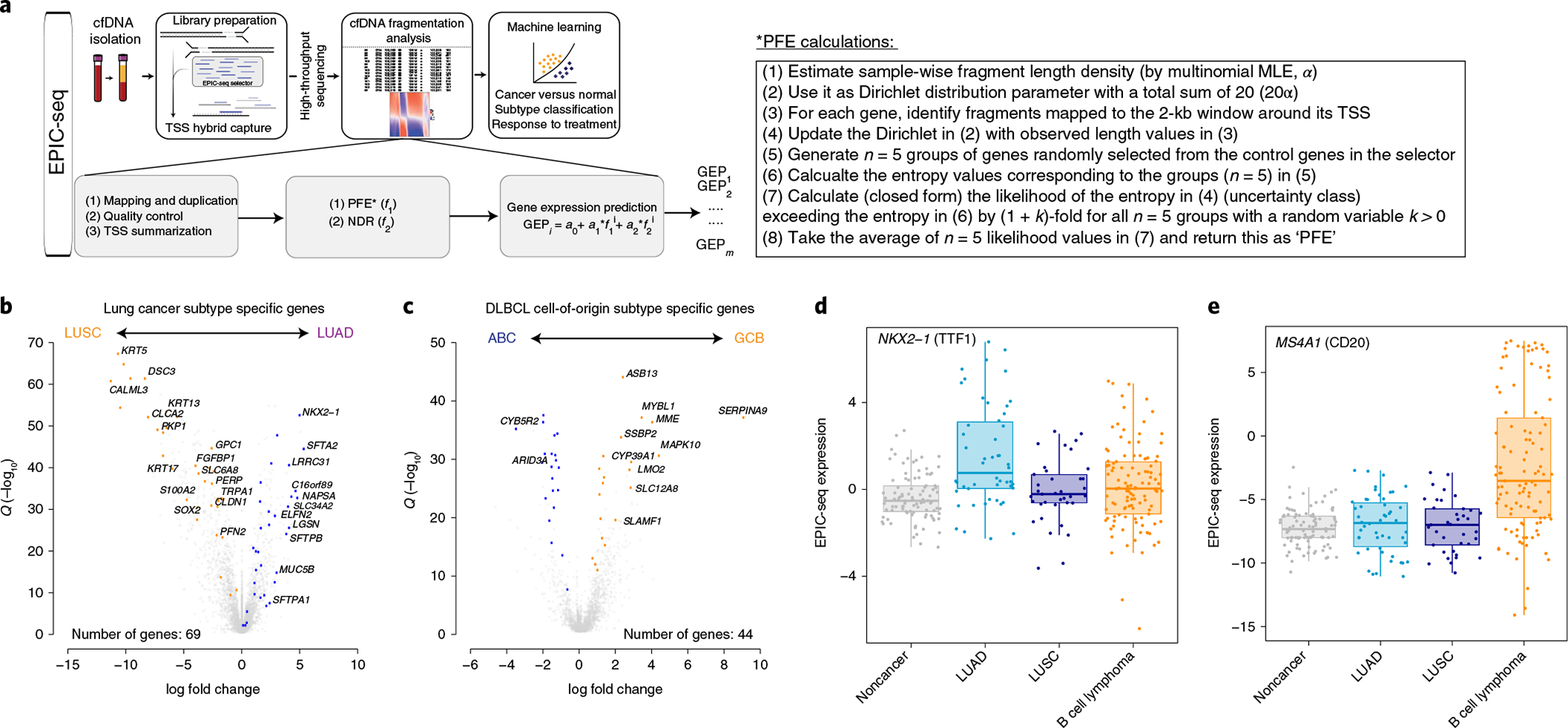
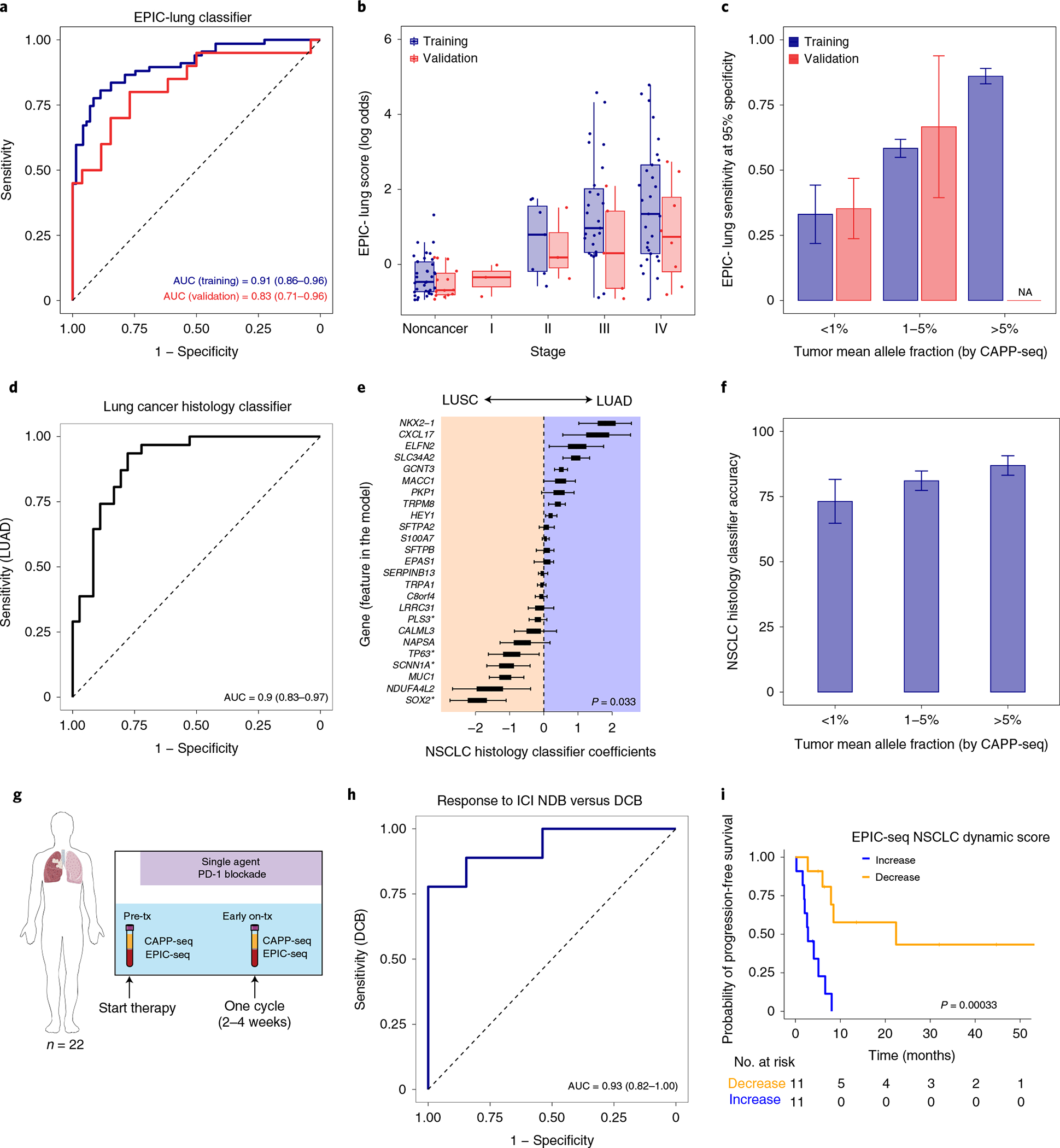
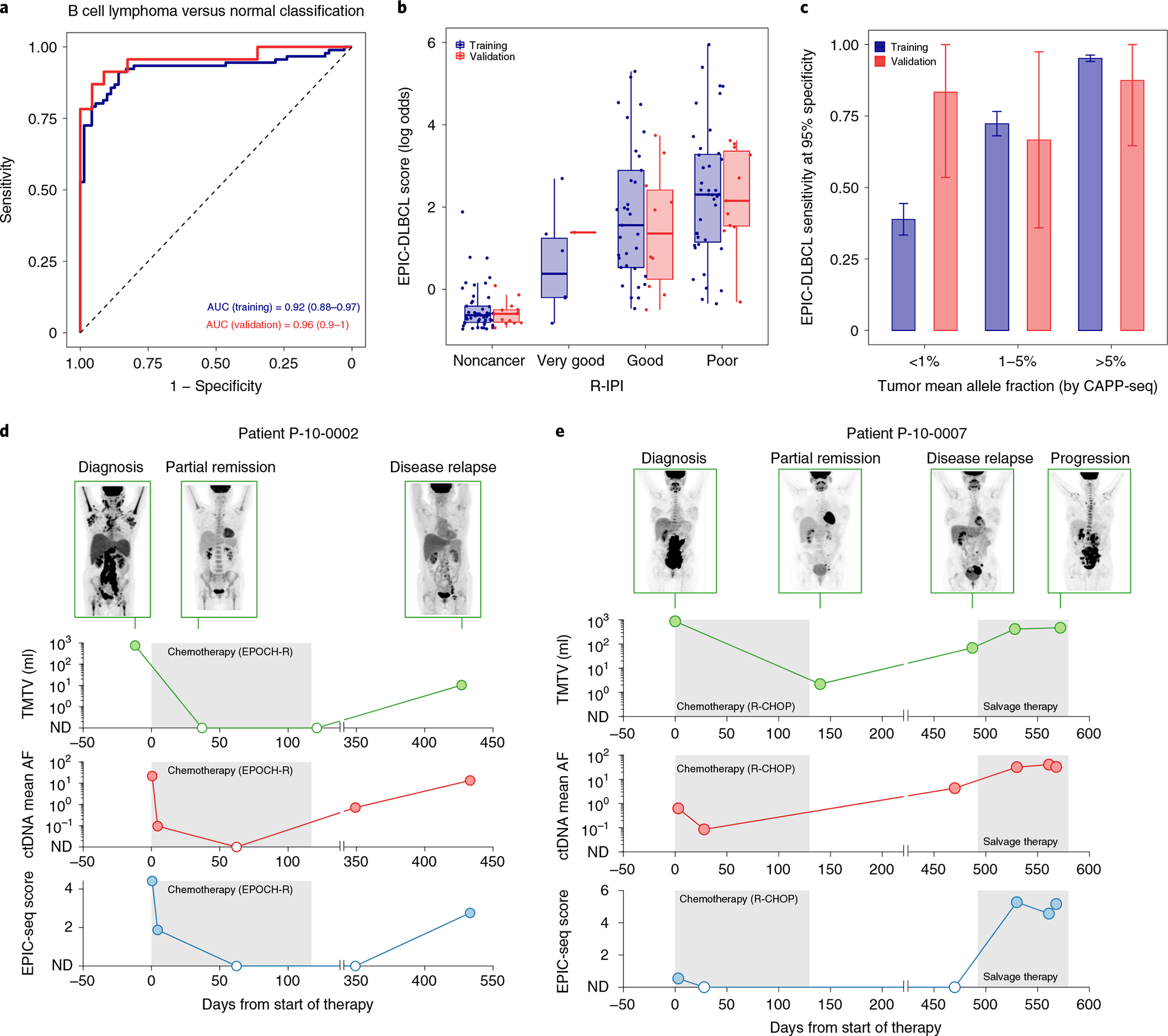
Profiling of circulating tumor DNA (ctDNA) in the bloodstream shows promise for noninvasive cancer detection. Chromatin fragmentation features have previously been explored to infer gene expression profiles from cell-free DNA (cfDNA), but current fragmentomic methods require high concentrations of tumor-derived DNA and provide limited resolution. Here we describe promoter fragmentation entropy as an epigenomic cfDNA feature that predicts RNA expression levels at individual genes. We developed 'epigenetic expression inference from cell-free DNA-sequencing' (EPIC-seq), a method that uses targeted sequencing of promoters of genes of interest. Profiling 329 blood samples from 201 patients with cancer and 87 healthy adults, we demonstrate classification of subtypes of lung carcinoma and diffuse large B cell lymphoma. Applying EPIC-seq to serial blood samples from patients treated with PD-(L)1 immune-checkpoint inhibitors, we show that gene expression profiles inferred by EPIC-seq are correlated with clinical response. Our results indicate that EPIC-seq could enable noninvasive, high-throughput tissue-of-origin characterization with diagnostic, prognostic and therapeutic potential.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
A.A.A. reports research funding from Celgene, Pfizer, ownership interests in FortySeven, CiberMed, ForeSight and paid consultancy from Roche, Genentech, Janssen, Pharmacyclics, Gilead, Celgene and Chugai. M.D. reports research funding from Varian Medical Systems and Illumina, ownership interest in CiberMed, ForeSight and paid consultancy from Roche, AstraZeneca, Illumina, RefleXion and BioNTech. J.J.C. reports paid consultancy from Lexent Bio Inc. and ownership interests in ForeSight. A.M.N. has patent filings related to expression deconvolution and cancer biomarkers and has served as a consultant for Roche, Merck and CiberMed. D.M.K. reports paid consultancy from Roche. B.T.L. has served as an uncompensated advisor and consultant to Amgen, Genentech, Boehringer Ingelheim, Lilly, AstraZeneca and Daiichi Sankyo. B.T.L. reports receiving research grants to his institution from Amgen, Genentech, AstraZeneca, Daiichi Sankyo, Lilly, Illumina, GRAIL, Guardant Health, Hengrui Therapeutics, MORE Health and Bolt Biotherapeutics. B.T.L. has received academic travel support from MORE Health and Jiangsu Hengrui Medicine. B.T.L. reports to be inventor on two institutional patents at MSKCC (US62/685,057, US62/514,661) and has intellectual property rights as a book author at Karger Publishers and Shanghai Jiao Tong University Press. J.M.I. reports serving as an unpaid consultant to Amgen and Roche-Genentech, institutional research support from Guardant Health and GRAIL, and ownership interest in LumaCyte. A.A.A., M.D., M.S.E., D.M.K., J.J.C., and B.Y.N. report patent filings related to cancer biomarkers. M.S.E., M.M., A.A.A. and M.D. have patent filing related to this paper. B.Y.N. is currently an employee and holds stock from Roche/Genentech. The remaining authors declare no competing interests.
Figures
Comment in
-
Enhanced cancer detection from cell-free DNA.Nat Biotechnol. 2022 Apr;40(4):473-474. doi: 10.1038/s41587-021-01207-9. Nat Biotechnol. 2022. PMID: 35361997 No abstract available.
-
Cell-free DNA cues for gene expression.Nat Methods. 2022 May;19(5):519. doi: 10.1038/s41592-022-01503-5. Nat Methods. 2022. PMID: 35545709 No abstract available.
References
-
- Jahr S et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 61, 1659–1665 (2001). - PubMed
-
- Lo YM et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2, 61ra91 (2010). - PubMed
-
- Heitzer E, Auinger L & Speicher MR Cell-free DNA and apoptosis: how dead cells inform about the living. Trends Mol. Med. 26, 519–528 (2020). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
