

The histone variant macroH2A1.1 regulates RNA polymerase II-paused genes within defined chromatin interaction landscapes
- PMID: 35362516
- PMCID: PMC9016624
- DOI: 10.1242/jcs.259456
The histone variant macroH2A1.1 regulates RNA polymerase II-paused genes within defined chromatin interaction landscapes
Abstract
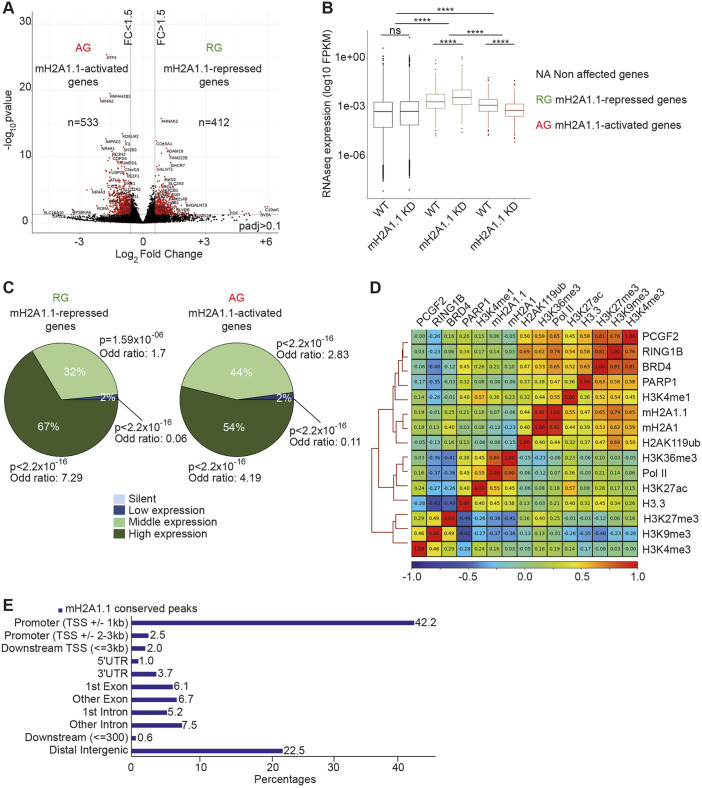
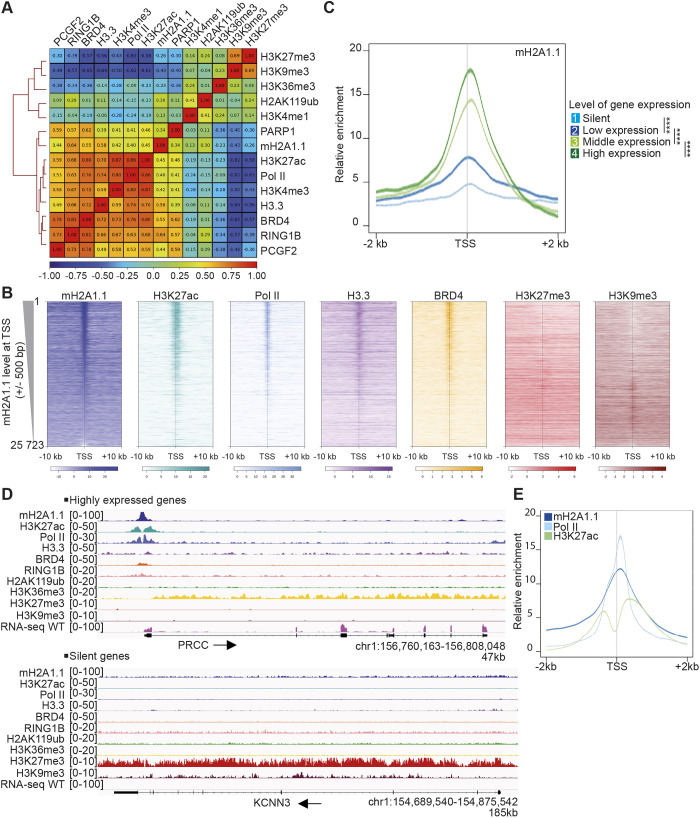
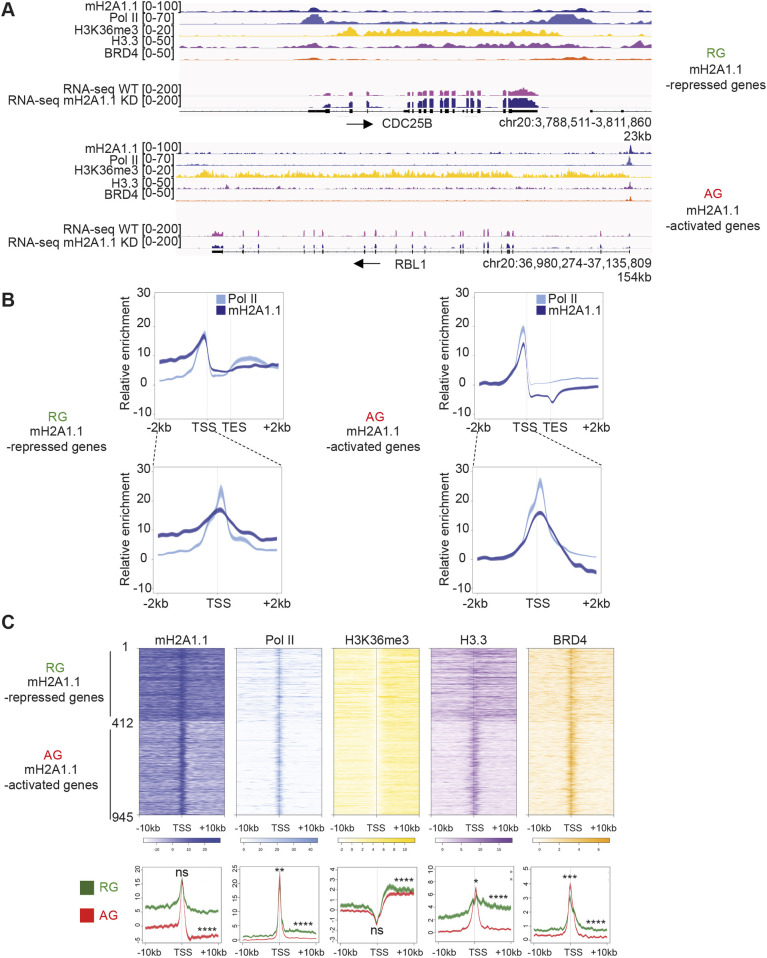
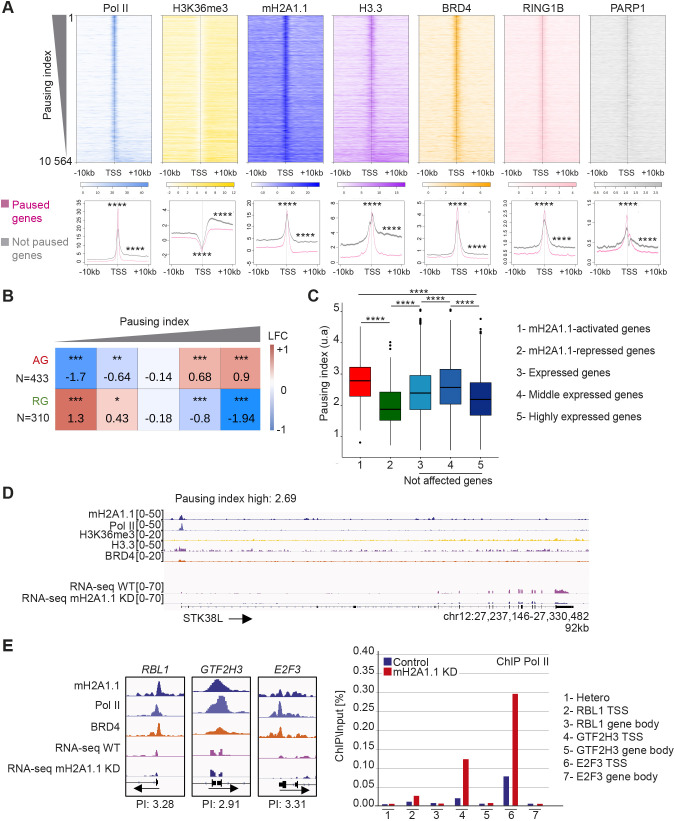
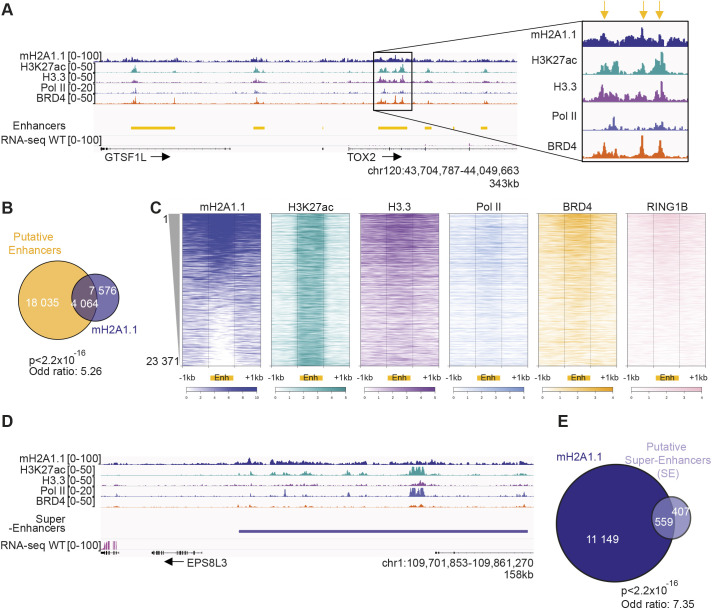
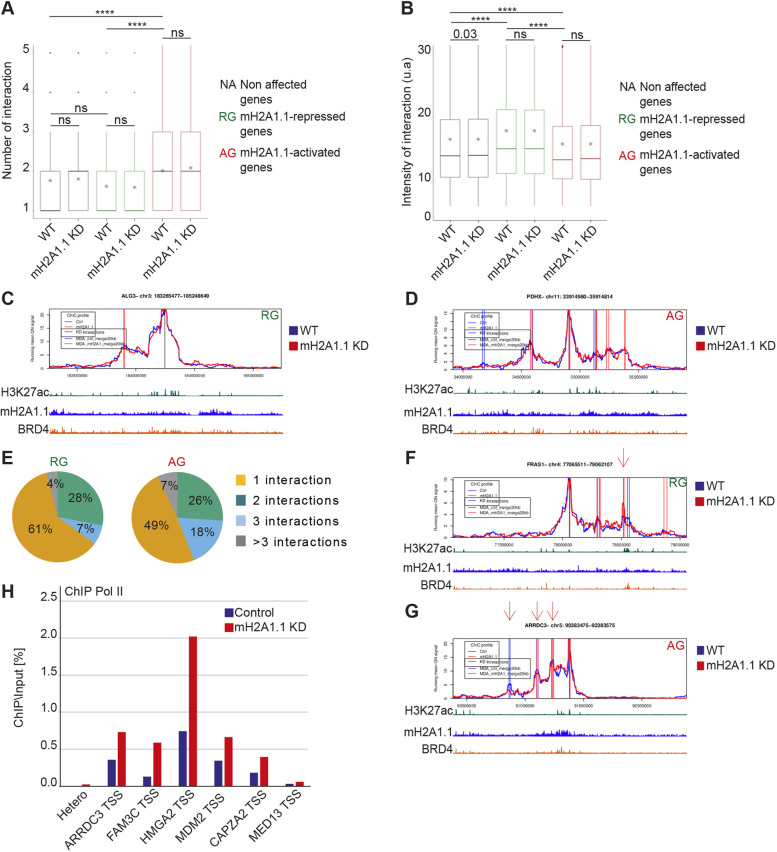
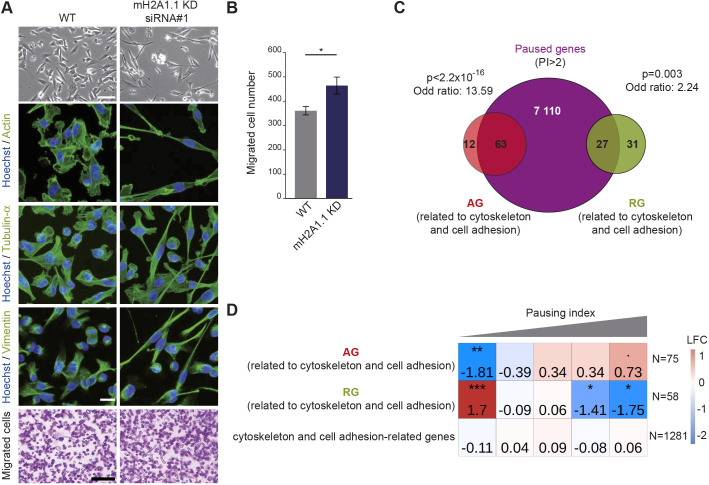
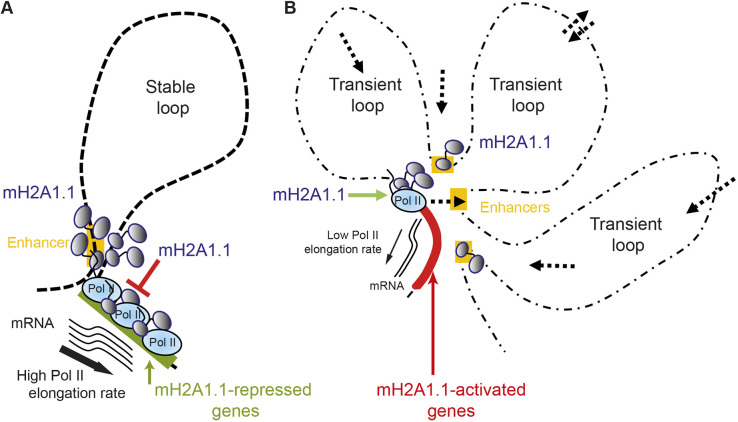
The histone variant macroH2A1.1 plays a role in cancer development and metastasis. To determine the underlying molecular mechanisms, we mapped the genome-wide localization of endogenous macroH2A1.1 in the human breast cancer cell line MDA-MB-231. We demonstrate that macroH2A1.1 specifically binds to active promoters and enhancers in addition to facultative heterochromatin. Selective knock down of macroH2A1.1 deregulates the expression of hundreds of highly active genes. Depending on the chromatin landscape, macroH2A1.1 acts through two distinct molecular mechanisms. The first mitigates excessive transcription by binding over domains including the promoter and the gene body. The second stimulates expression of RNA polymerase II (Pol II)-paused genes, including genes regulating mammary tumor cell migration. In contrast to the first mechanism, macroH2A1.1 specifically associates with the transcription start site of Pol II-paused genes. These processes occur in a predefined local 3D genome landscape, but do not require rewiring of enhancer-promoter contacts. We thus propose that macroH2A1.1 serves as a transcriptional modulator with a potential role in assisting the conversion of promoter-locked Pol II into a productive, elongating Pol II.
Keywords: Breast cancer; Cellular migration; Chromatin structure; Gene expression; Histone variants; RNA polymerase II-paused genes.
© 2022. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interests The authors declare no competing or financial interests.
Figures
References
-
- Bejjani, F., Tolza, C., Boulanger, M., Downes, D., Romero, R., Maqbool, M. A., Zine El Aabidine, A., Andrau, J.-C., Lebre, S., Brehelin, L.et al. (2021). Fra-1 regulates its target genes via binding to remote enhancers without exerting major control on chromatin architecture in triple negative breast cancers. Nucleic Acids Res. 49, 2488-2508. 10.1093/nar/gkab053 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
