

Deubiquitinase CYLD acts as a negative regulator of dopamine neuron survival in Parkinson's disease
- PMID: 35363524
- PMCID: PMC10938605
- DOI: 10.1126/sciadv.abh1824
Deubiquitinase CYLD acts as a negative regulator of dopamine neuron survival in Parkinson's disease
Abstract
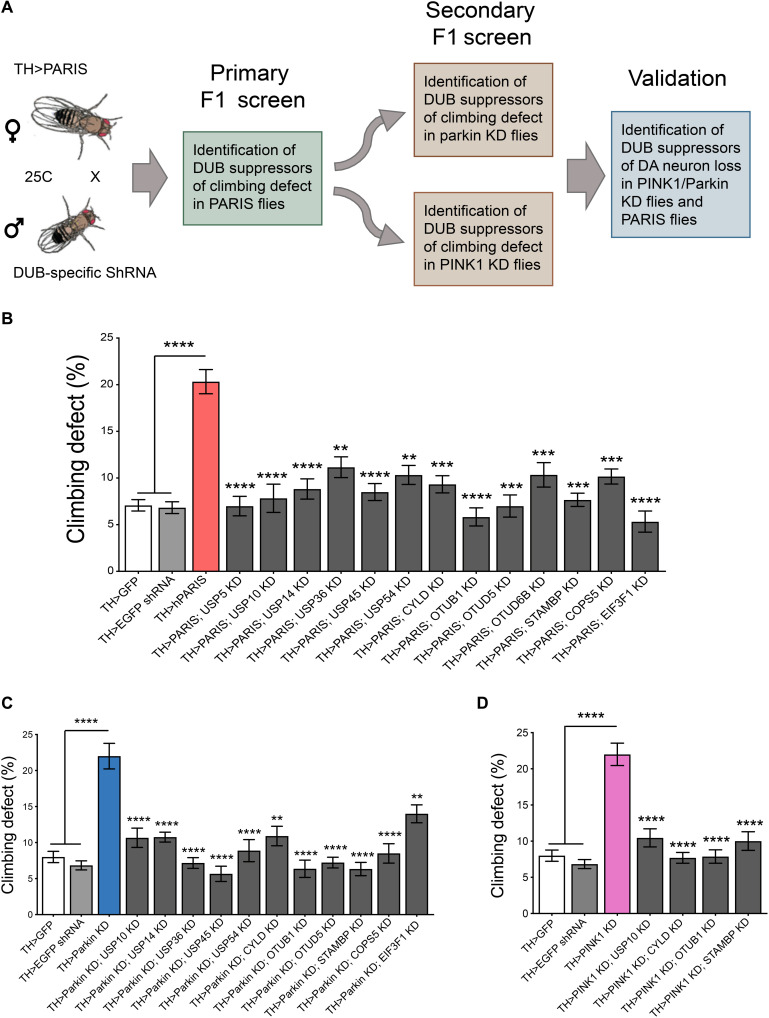
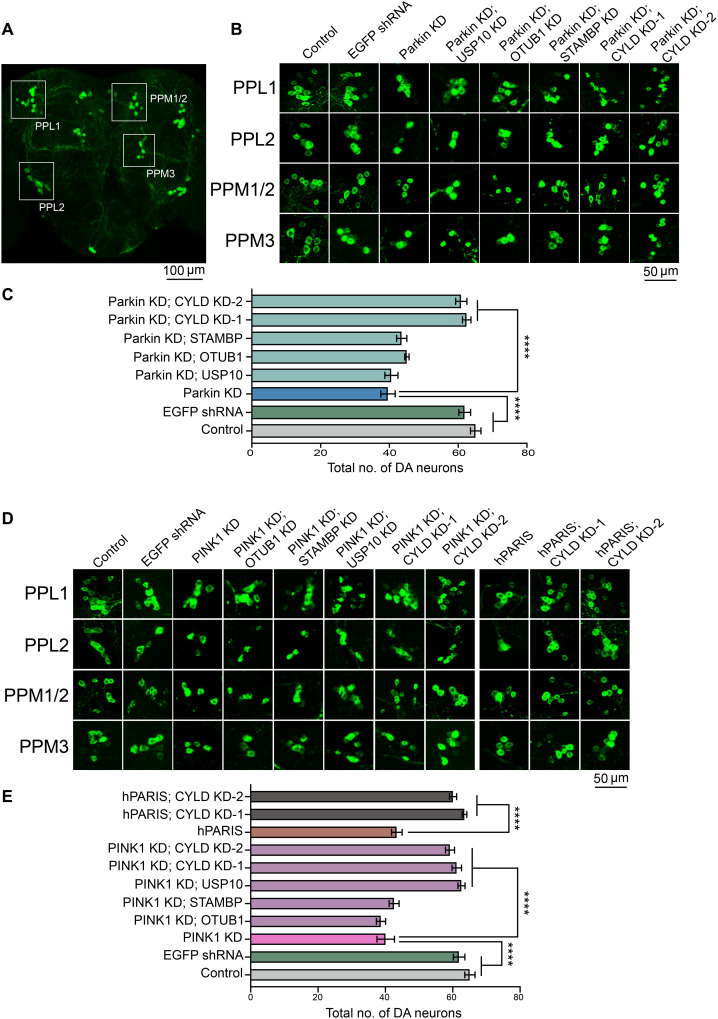
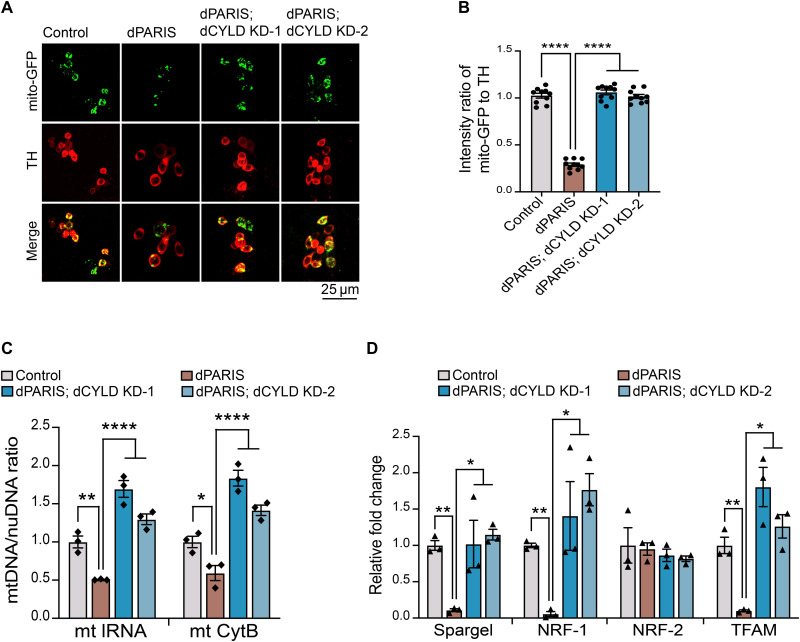
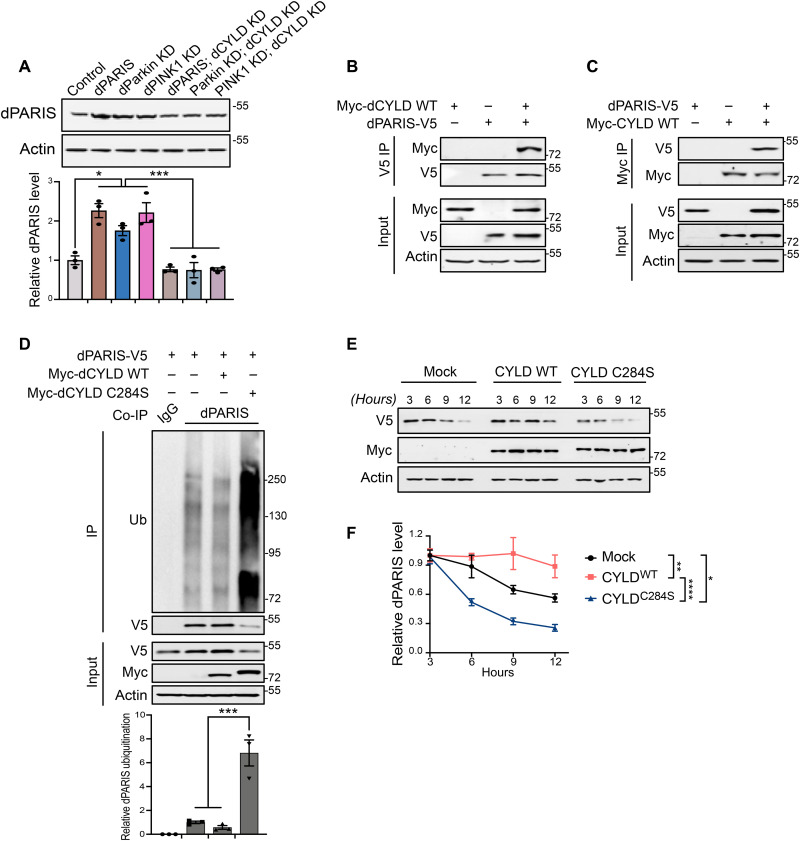
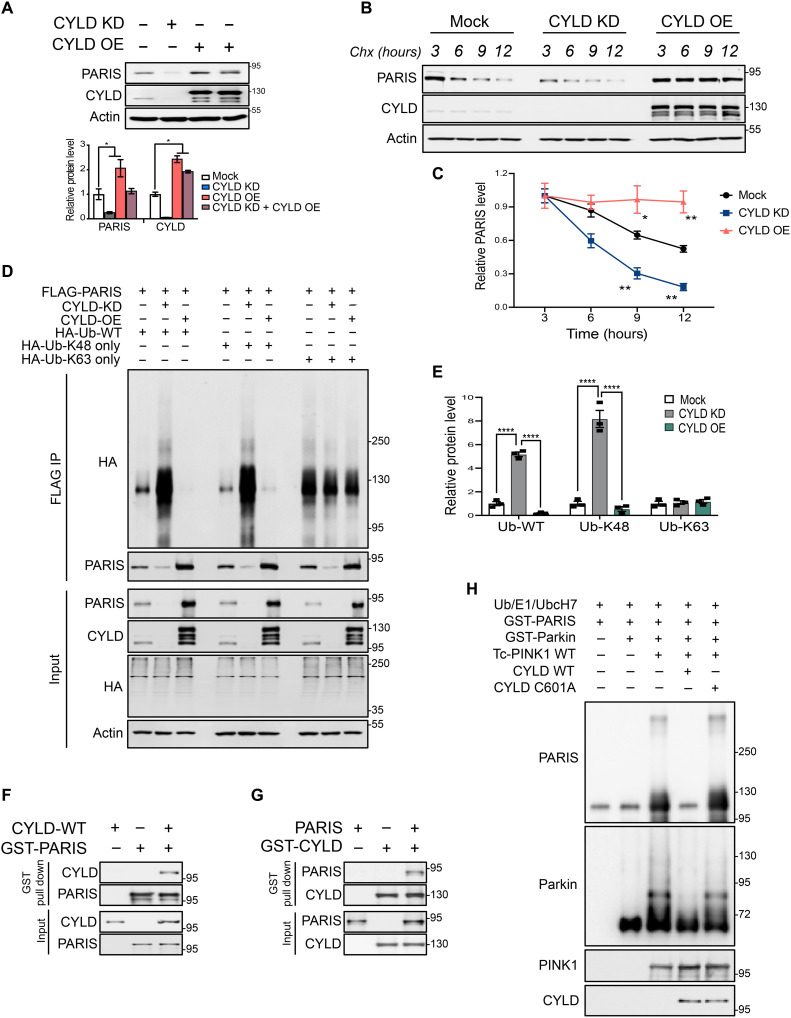
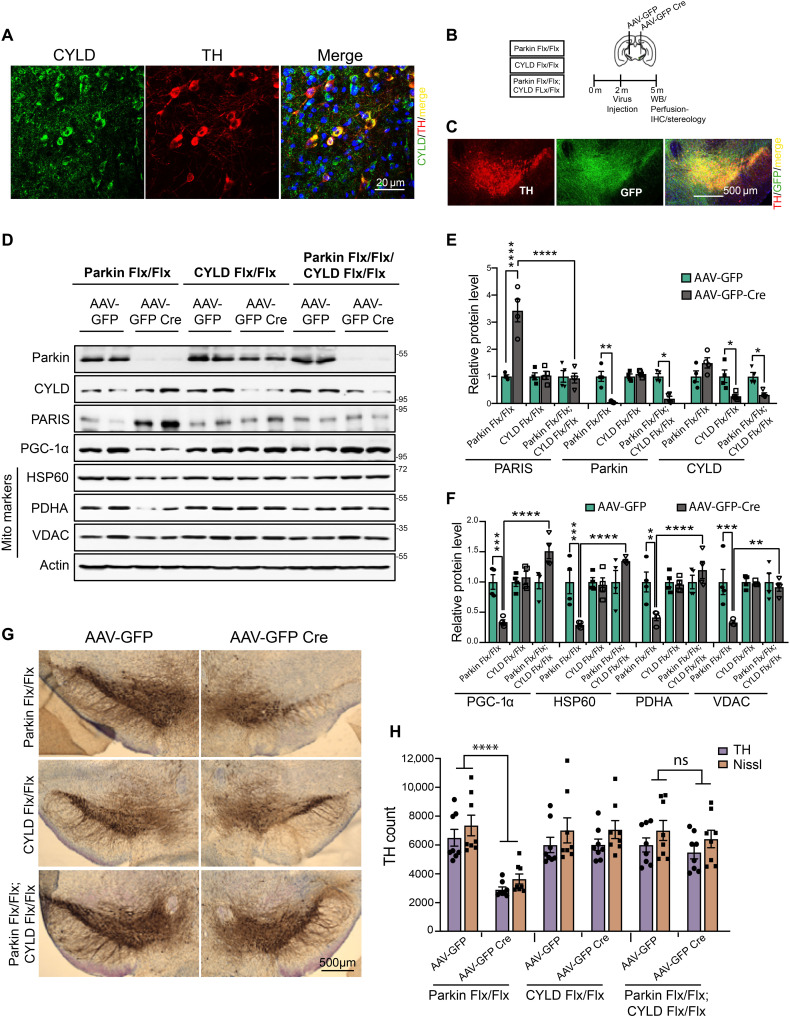
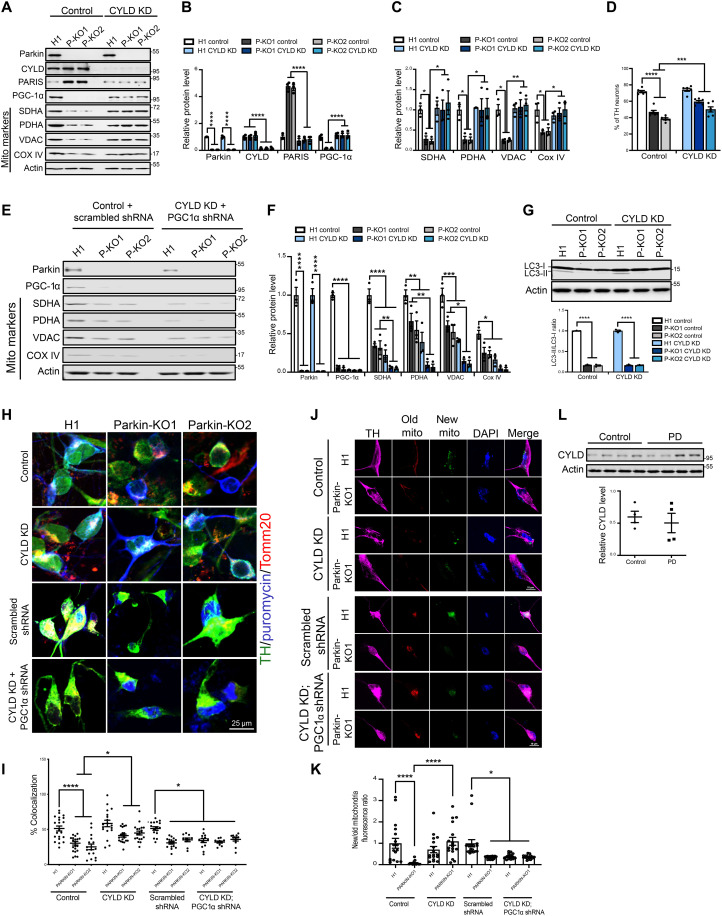
Mutations in PINK1 and parkin highlight the mitochondrial axis of Parkinson's disease (PD) pathogenesis. PINK1/parkin regulation of the transcriptional repressor PARIS bears direct relevance to dopamine neuron survival through augmentation of PGC-1α-dependent mitochondrial biogenesis. Notably, knockout of PARIS attenuates dopaminergic neurodegeneration in mouse models, indicating that interventions that prevent dopaminergic accumulation of PARIS could have therapeutic potential in PD. To this end, we have identified the deubiquitinase cylindromatosis (CYLD) to be a regulator of PARIS protein stability and proteasomal degradation via the PINK1/parkin pathway. Knockdown of CYLD in multiple models of PINK1 or parkin inactivation attenuates PARIS accumulation by modulating its ubiquitination levels and relieving its repressive effect on PGC-1α to promote mitochondrial biogenesis. Together, our studies identify CYLD as a negative regulator of dopamine neuron survival, and inhibition of CYLD may potentially be beneficial in PD by lowering PARIS levels and promoting mitochondrial biogenesis.
Figures
Similar articles
-
PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency.Mol Neurodegener. 2020 Mar 5;15(1):17. doi: 10.1186/s13024-020-00363-x. Mol Neurodegener. 2020. PMID: 32138754 Free PMC article.
-
PINK1 Primes Parkin-Mediated Ubiquitination of PARIS in Dopaminergic Neuronal Survival.Cell Rep. 2017 Jan 24;18(4):918-932. doi: 10.1016/j.celrep.2016.12.090. Cell Rep. 2017. PMID: 28122242 Free PMC article.
-
Preclinical studies and transcriptome analysis in a model of Parkinson's disease with dopaminergic ZNF746 expression.Mol Neurodegener. 2025 Feb 28;20(1):24. doi: 10.1186/s13024-025-00814-3. Mol Neurodegener. 2025. PMID: 40022229 Free PMC article.
-
[The role of parkin in Parkinson's disease].Neuropsychopharmacol Hung. 2014 Jun;16(2):67-76. Neuropsychopharmacol Hung. 2014. PMID: 24978049 Review. Hungarian.
-
Unraveling correlative roles of dopamine transporter (DAT) and Parkin in Parkinson's disease (PD) - A road to discovery?Brain Res Bull. 2020 Apr;157:169-179. doi: 10.1016/j.brainresbull.2020.02.001. Epub 2020 Feb 6. Brain Res Bull. 2020. PMID: 32035946 Review.
Cited by
-
Cell Biology of Parkin: Clues to the Development of New Therapeutics for Parkinson's Disease.CNS Drugs. 2022 Dec;36(12):1249-1267. doi: 10.1007/s40263-022-00973-7. Epub 2022 Nov 15. CNS Drugs. 2022. PMID: 36378485 Review.
-
Investigating the interplay between mitophagy and diabetic neuropathy: Uncovering the hidden secrets of the disease pathology.Pharmacol Res. 2024 Oct;208:107394. doi: 10.1016/j.phrs.2024.107394. Epub 2024 Sep 3. Pharmacol Res. 2024. PMID: 39233055 Review.
-
Bioactivated Glucoraphanin Modulates Genes Involved in Necroptosis on Motor-Neuron-like Nsc-34: A Transcriptomic Study.Antioxidants (Basel). 2024 Sep 14;13(9):1111. doi: 10.3390/antiox13091111. Antioxidants (Basel). 2024. PMID: 39334770 Free PMC article.
-
The Potential of Cylindromatosis (CYLD) as a Therapeutic Target in Oxidative Stress-Associated Pathologies: A Comprehensive Evaluation.Int J Mol Sci. 2023 May 6;24(9):8368. doi: 10.3390/ijms24098368. Int J Mol Sci. 2023. PMID: 37176077 Free PMC article. Review.
-
USP14 inhibits mitophagy and promotes tumorigenesis and chemosensitivity through deubiquitinating BAG4 in microsatellite instability-high colorectal cancer.Mol Med. 2025 May 2;31(1):163. doi: 10.1186/s10020-025-01182-w. Mol Med. 2025. PMID: 40316942 Free PMC article.
References
-
- Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N., Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 (1998). - PubMed
-
- Valente E. M., Abou-Sleiman P. M., Caputo V., Muqit M. M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A. R., Healy D. G., Albanese A., Nussbaum R., Gonzalez-Maldonado R., Deller T., Salvi S., Cortelli P., Gilks W. P., Latchman D. S., Harvey R. J., Dallapiccola B., Auburger G., Wood N. W., Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160 (2004). - PubMed
-
- Koyano F., Okatsu K., Kosako H., Tamura Y., Go E., Kimura M., Kimura Y., Tsuchiya H., Yoshihara H., Hirokawa T., Endo T., Fon E. A., Trempe J. F., Saeki Y., Tanaka K., Matsuda N., Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166 (2014). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
