

Protective role of chaperone-mediated autophagy against atherosclerosis
- PMID: 35363568
- PMCID: PMC9168839
- DOI: 10.1073/pnas.2121133119
Protective role of chaperone-mediated autophagy against atherosclerosis
Abstract
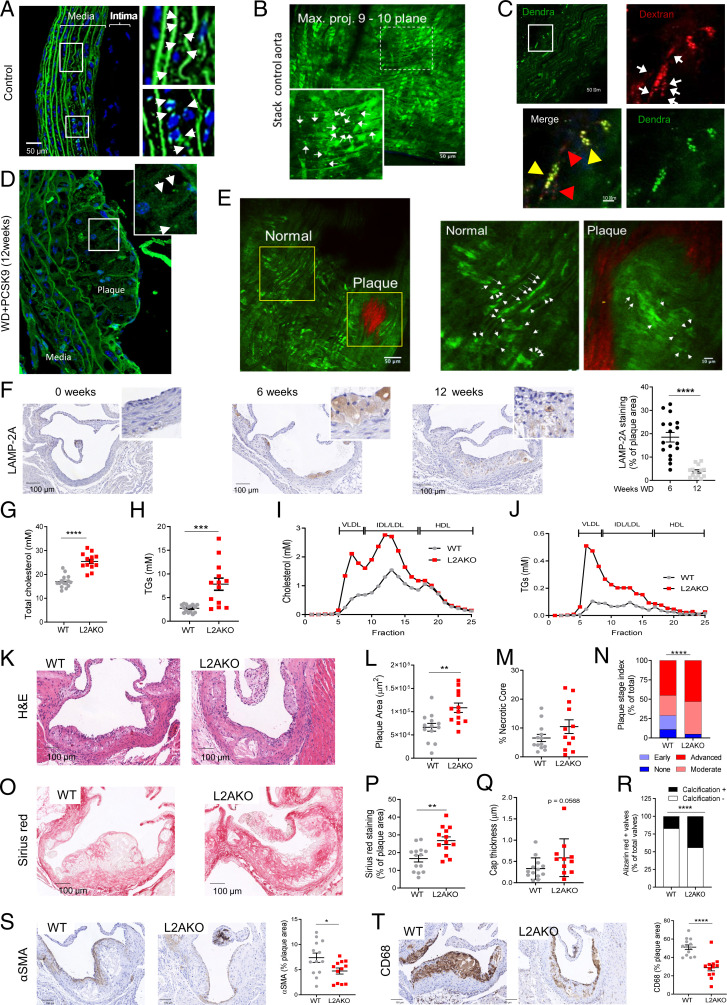
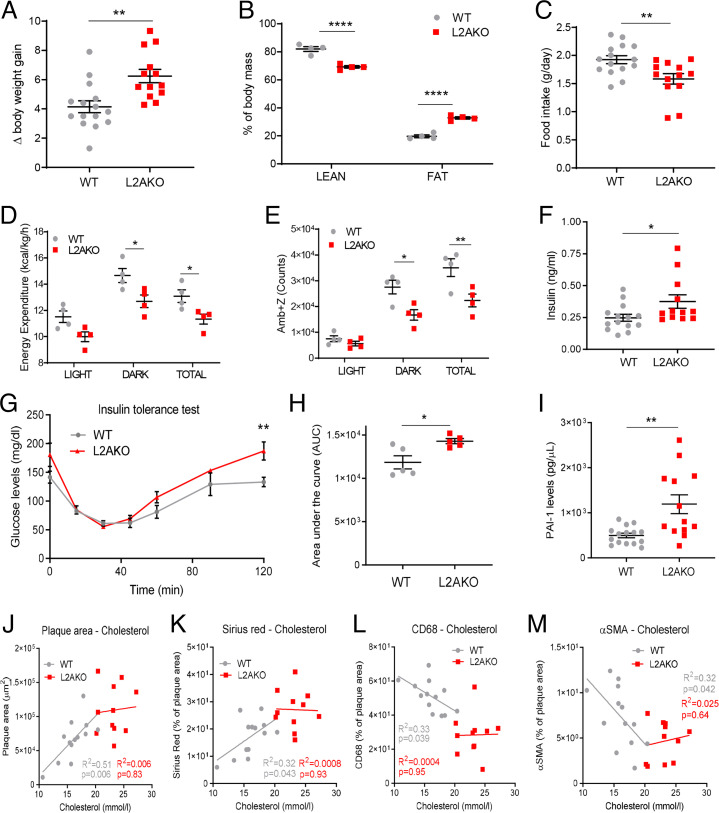
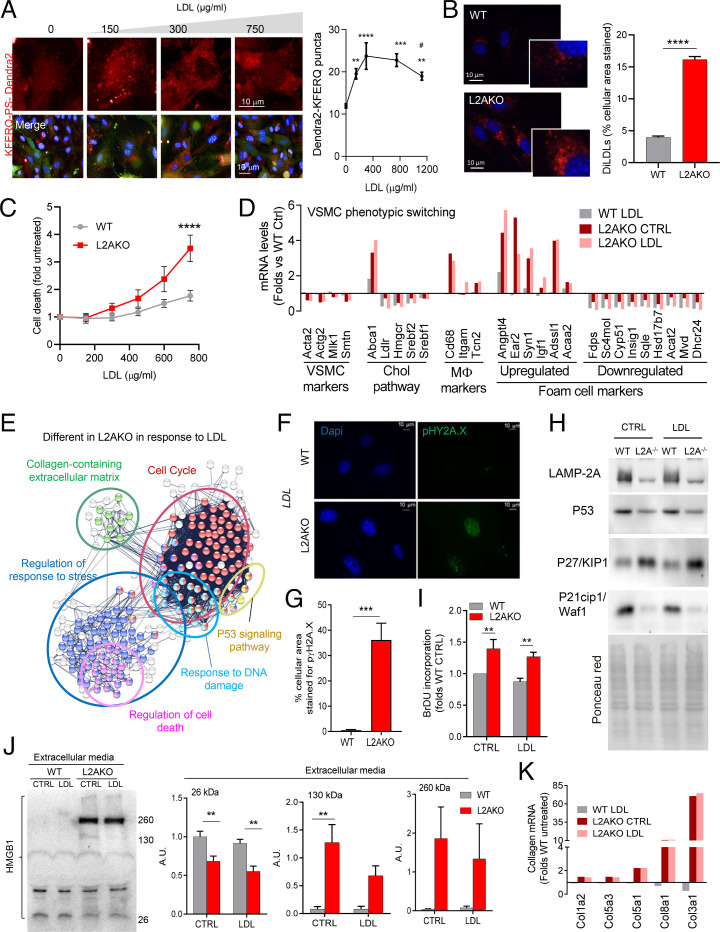
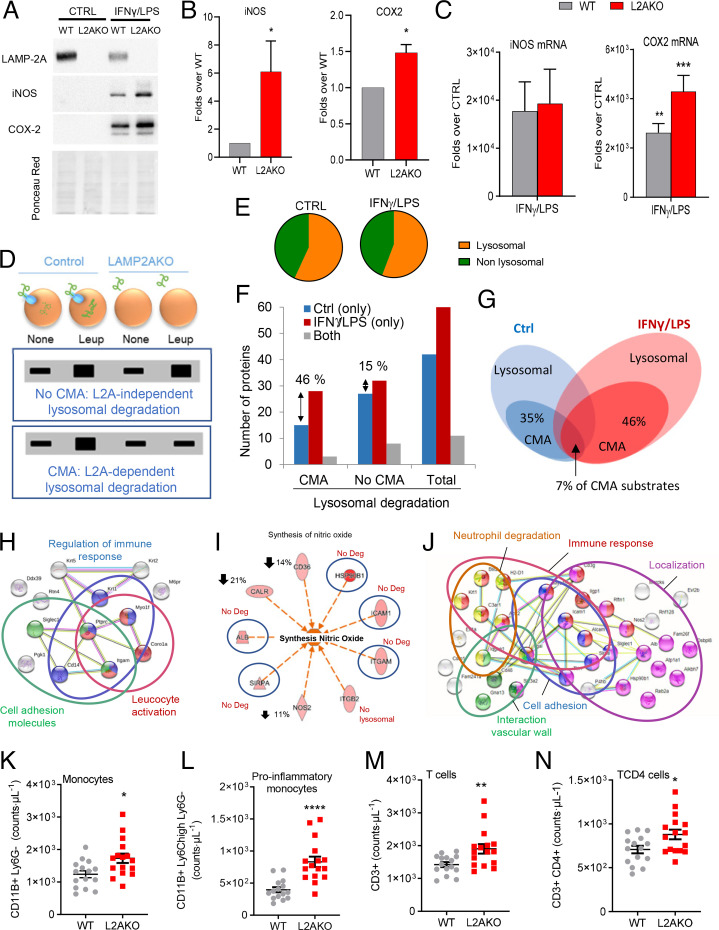
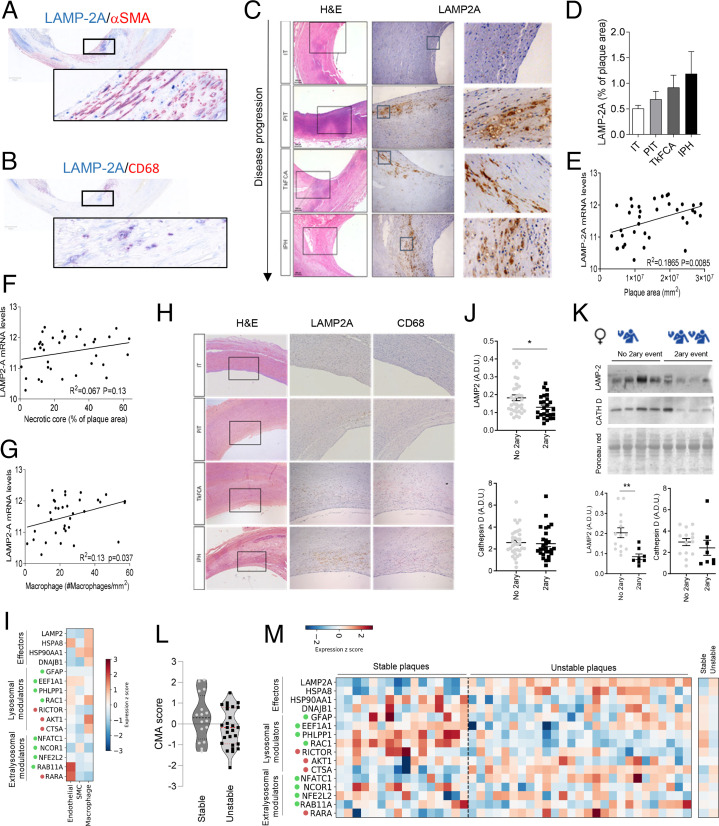
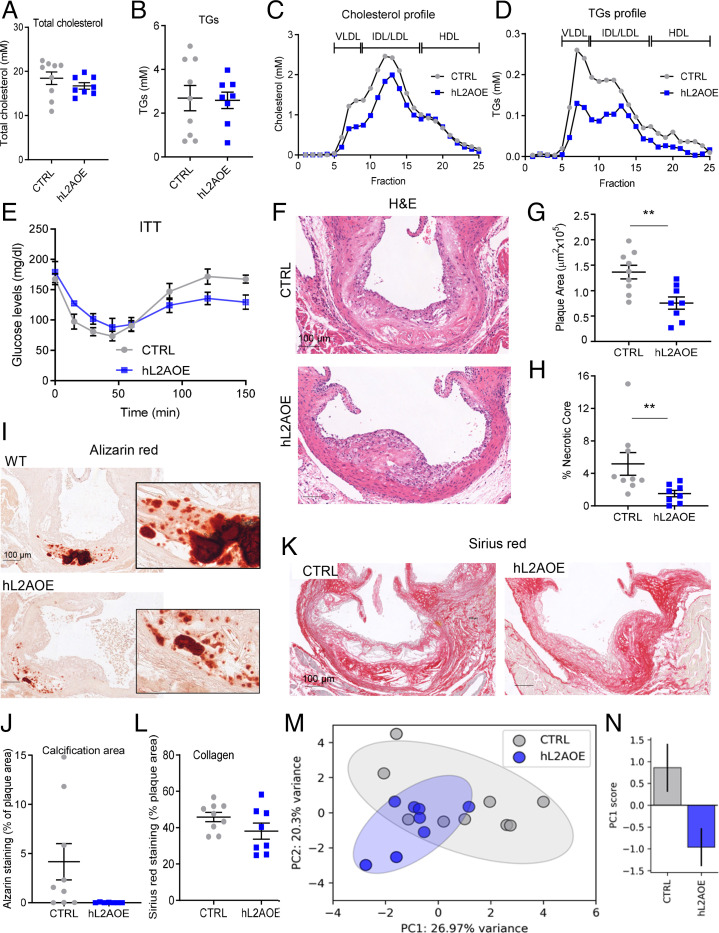
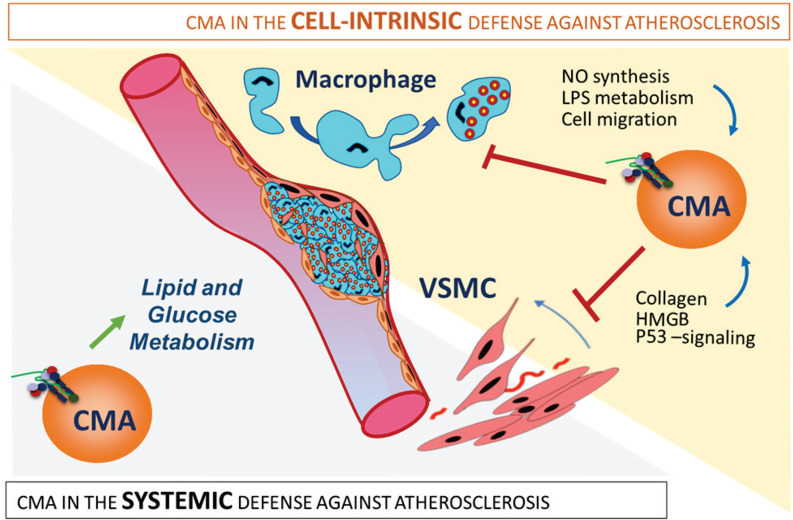
Chaperone-mediated autophagy (CMA) contributes to regulation of energy homeostasis by timely degradation of enzymes involved in glucose and lipid metabolism. Here, we report reduced CMA activity in vascular smooth muscle cells and macrophages in murine and human arteries in response to atherosclerotic challenges. We show that in vivo genetic blockage of CMA worsens atherosclerotic pathology through both systemic and cell-autonomous changes in vascular smooth muscle cells and macrophages, the two main cell types involved in atherogenesis. CMA deficiency promotes dedifferentiation of vascular smooth muscle cells and a proinflammatory state in macrophages. Conversely, a genetic mouse model with up-regulated CMA shows lower vulnerability to proatherosclerotic challenges. We propose that CMA could be an attractive therapeutic target against cardiovascular diseases.
Keywords: atherosclerotic plaques; lipid challenge; lysosomes; proteolysis; vascular disease.
Conflict of interest statement
Competing interest statement: J.M.-M. is a founder and consultant for Instituto Ibions SLP. A.H.B. is a coauthor with T.F. of a review article in 2019. A.M.C. is a cofounder and scientific advisor for the autophagy program at Life Biosciences and consults for Generian Pharmaceuticals and Cognition Therapeutics.
Figures
Comment in
-
Chaperone-mediated autophagy protects against atherosclerosis.Autophagy. 2022 Oct;18(10):2505-2507. doi: 10.1080/15548627.2022.2096397. Epub 2022 Jul 19. Autophagy. 2022. PMID: 35787098 Free PMC article.
References
-
- Virani S. S., et al. ; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee, Heart disease and stroke statistics—2021 update: A report from the American Heart Association. Circulation 143, e254–e743 (2021). - PubMed
-
- Libby P., The changing landscape of atherosclerosis. Nature 592, 524–533 (2021). - PubMed
-
- Tabas I., Williams K. J., Borén J., Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 116, 1832–1844 (2007). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
