

Molecular subclusters of follicular lymphoma: a report from the United Kingdom's Haematological Malignancy Research Network
- PMID: 35363872
- PMCID: PMC9619185
- DOI: 10.1182/bloodadvances.2021005284
Molecular subclusters of follicular lymphoma: a report from the United Kingdom's Haematological Malignancy Research Network
Abstract
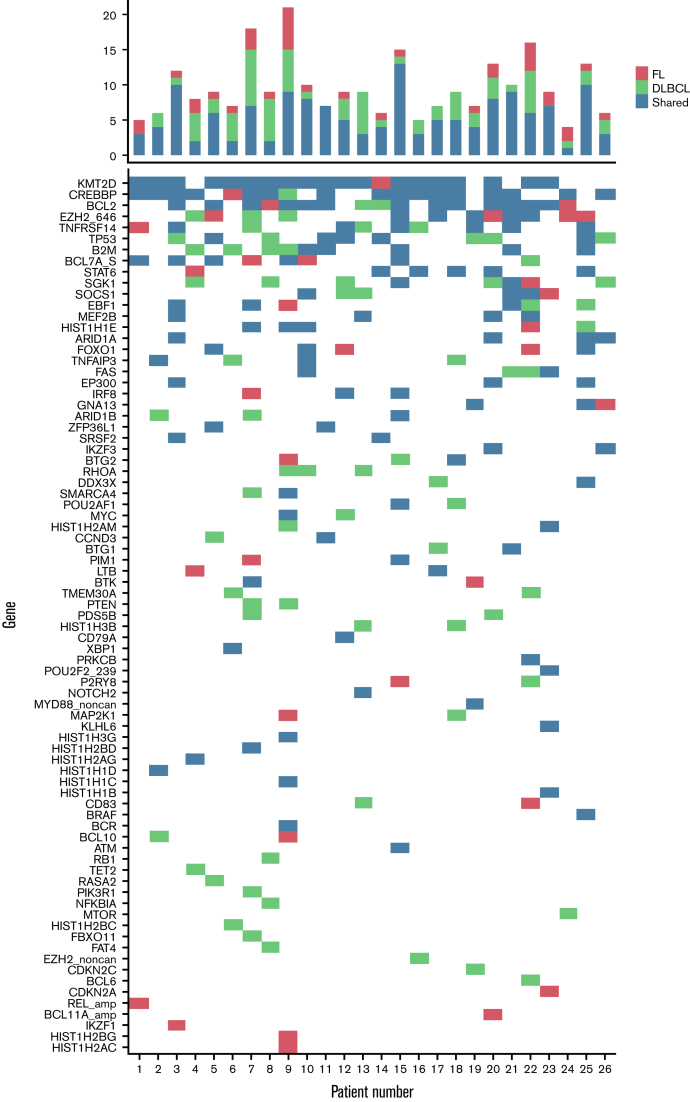
Follicular lymphoma (FL) is morphologically and clinically diverse, with mutations in epigenetic regulators alongside t(14;18) identified as disease-initiating events. Identification of additional mutational entities confirms this cancer's heterogeneity, but whether mutational data can be resolved into mechanistically distinct subsets remains an open question. Targeted sequencing was applied to an unselected population-based FL cohort (n = 548) with full clinical follow-up (n = 538), which included 96 diffuse large B-cell lymphoma (DLBCL) transformations. We investigated whether molecular subclusters of FL can be identified and whether mutational data provide predictive information relating to transformation. DNA extracted from FL samples was sequenced with a 293-gene panel representing genes frequently mutated in DLBCL and FL. Three clusters were resolved using mutational data alone, independent of translocation status: FL_aSHM, with high burden of aberrant somatic hypermutation (aSHM) targets; FL_STAT6, with high STAT6 & CREBBP mutation and low aSHM; and FL_Com, with the absence of features of other subtypes and enriched KMT2D mutation. Analysis of mutation signatures demonstrated differential enrichment of predicted mutation signatures between subgroups and a dominant preference in the FL_aSHM subgroup for G(C>T)T and G(C>T)C transitions consistent with previously defined aSHM-like patterns. Of transformed cases with paired samples, 17 of 26 had evidence of branching evolution. Poorer overall survival (OS) in the aSHM group (P = .04) was associated with older age; however, overall tumor genetics provided limited information to predict individual patient risk. Our approach identifies 3 molecular subclusters of FL linked to differences in underlying mechanistic pathways. These clusters, which may be further resolved by the inclusion of translocation status and wider mutation profiles, have implications for understanding pathogenesis as well as improving treatment strategies in the future.
© 2022 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: P.A.B. is the director at Tessellex Ltd and Gabriel Precision Oncology Ltd. D.J.H. receives research funding from Gilead International and provides consultancy for Karus. The remaining authors declare no competing financial interests.
The current affiliations for C.R. are the Department of Bioengineering and the Department of Computer Science, Stanford University, Palo Alto, CA. P.A.B. is currently affiliated with Hull York Medical School, Heslington, York.
Figures







References
-
- Basso K, Dalla-Favera R. Germinal centres and B cell lymphomagenesis. Nat Rev Immunol. 2015;15(3):172–184. - PubMed
-
- Basso K, Dalla-Favera R. BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv Immunol. 2010;105:193–210. - PubMed
-
- Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228(4706):1440–1443. - PubMed
-
- Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce CM. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science. 1985;229(4720):1390–1393. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
