

The rise of genomics in snake venom research: recent advances and future perspectives
- PMID: 35365832
- PMCID: PMC8975721
- DOI: 10.1093/gigascience/giac024
The rise of genomics in snake venom research: recent advances and future perspectives
Abstract
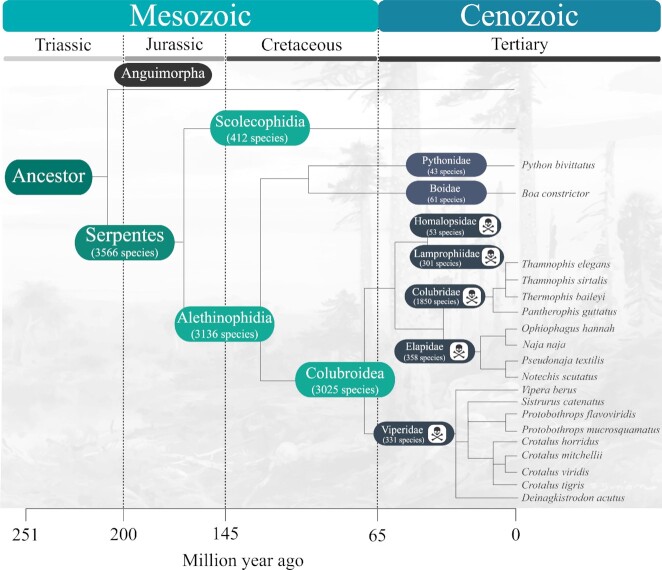
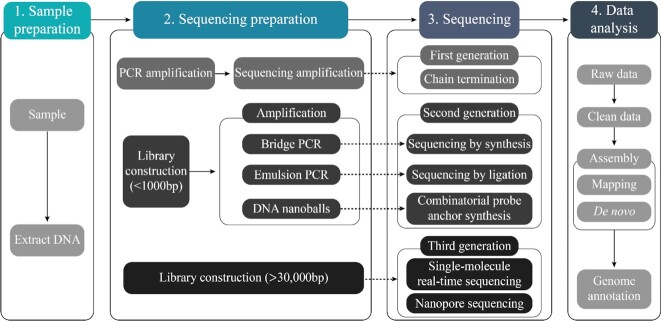
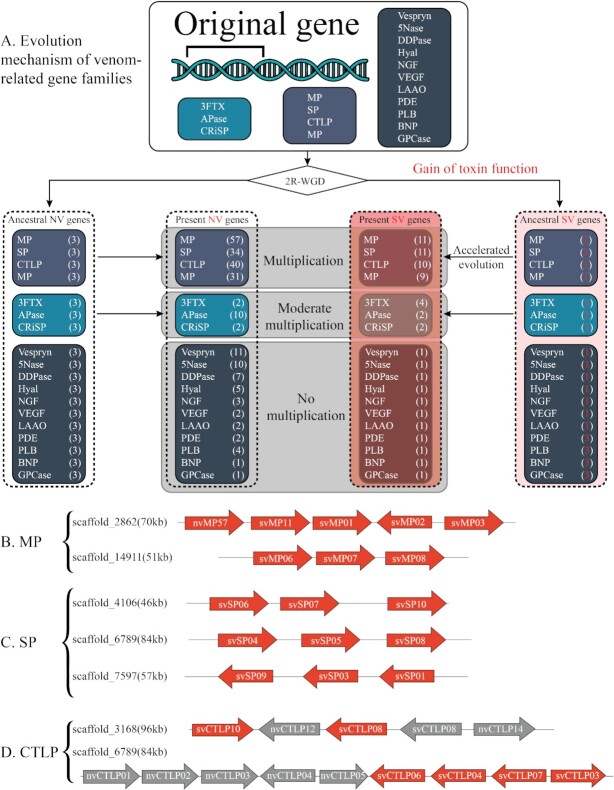
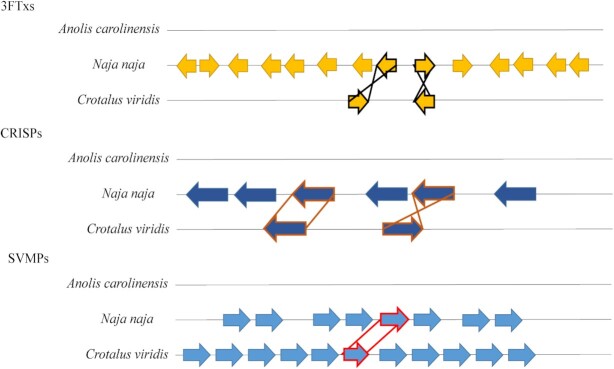
Snake venoms represent a danger to human health, but also a gold mine of bioactive proteins that can be harnessed for drug discovery purposes. The evolution of snakes and their venom has been studied for decades, particularly via traditional morphological and basic genetic methods alongside venom proteomics. However, while the field of genomics has matured rapidly over the past 2 decades, owing to the development of next-generation sequencing technologies, snake genomics remains in its infancy. Here, we provide an overview of the state of the art in snake genomics and discuss its potential implications for studying venom evolution and toxinology. On the basis of current knowledge, gene duplication and positive selection are key mechanisms in the neofunctionalization of snake venom proteins. This makes snake venoms important evolutionary drivers that explain the remarkable venom diversification and adaptive variation observed in these reptiles. Gene duplication and neofunctionalization have also generated a large number of repeat sequences in snake genomes that pose a significant challenge to DNA sequencing, resulting in the need for substantial computational resources and longer sequencing read length for high-quality genome assembly. Fortunately, owing to constantly improving sequencing technologies and computational tools, we are now able to explore the molecular mechanisms of snake venom evolution in unprecedented detail. Such novel insights have the potential to affect the design and development of antivenoms and possibly other drugs, as well as provide new fundamental knowledge on snake biology and evolution.
Keywords: DNA sequencing; snake genomics; snake toxins; snakes; venom; venom evolution.
© The Author(s) 2022. Published by Oxford University Press GigaScience.
Figures
References
-
- Burbrink FT, Pyron RA.. The taming of the skew: estimating proper confidence intervals for divergence dates. Syst Biol. 2008;57(2):317–28. - PubMed
-
- Wallach V, Williams KL, Boundy J.. Snakes of the World: A Catalogue of Living and Extinct Species. 1st ed. Boca Raton: CRC Press; 2014.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
