

The virome of the white-winged vampire bat Diaemus youngi is rich in circular DNA viruses
- PMID: 35366197
- PMCID: PMC8976263
- DOI: 10.1007/s11262-022-01897-6
The virome of the white-winged vampire bat Diaemus youngi is rich in circular DNA viruses
Abstract
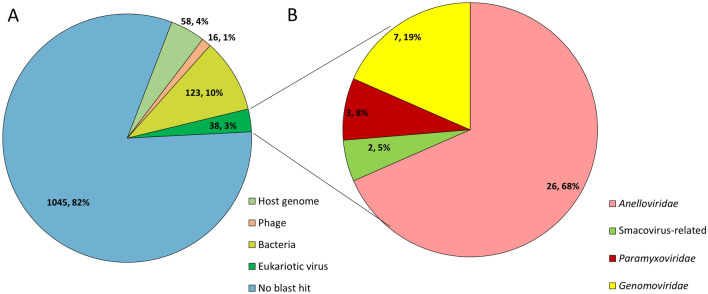
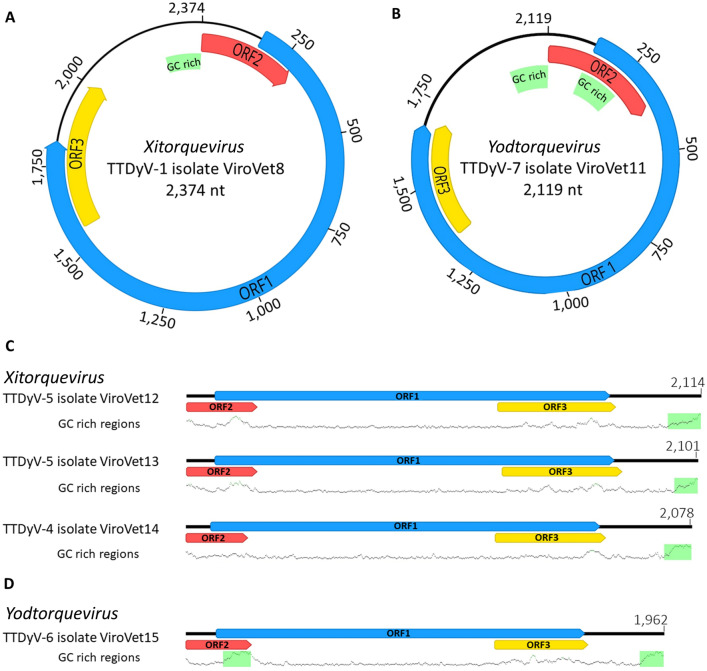
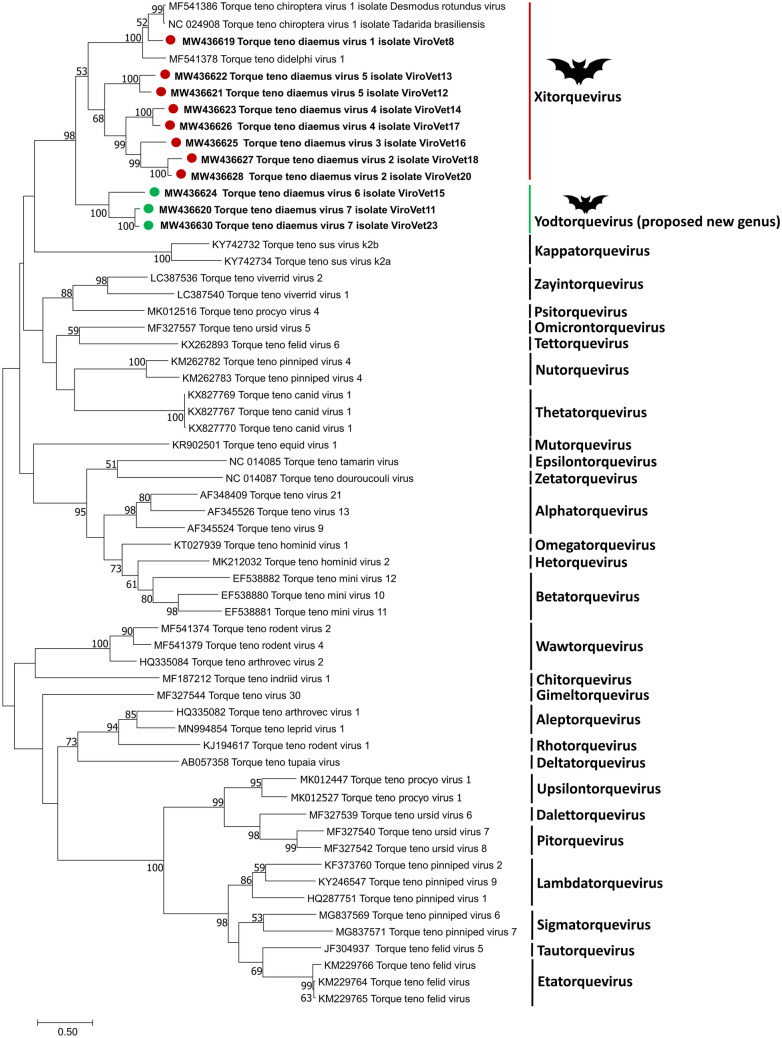
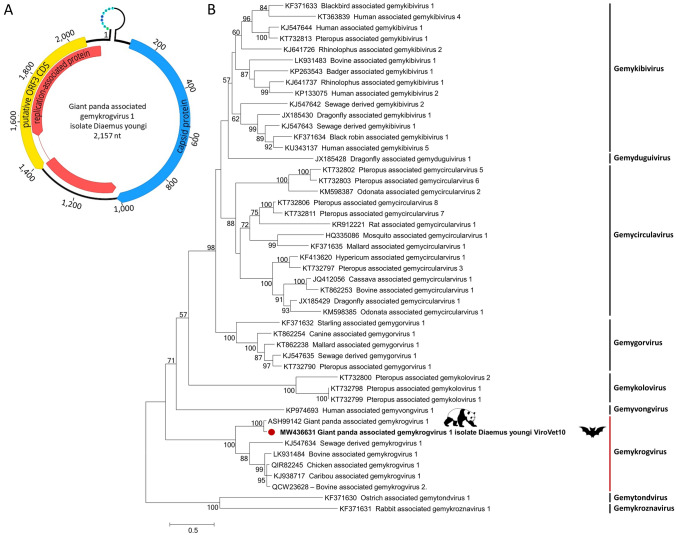
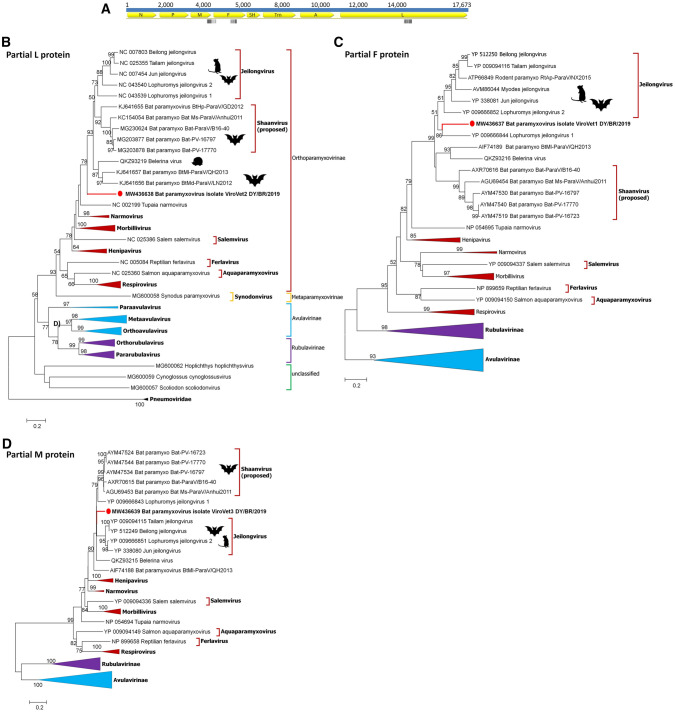
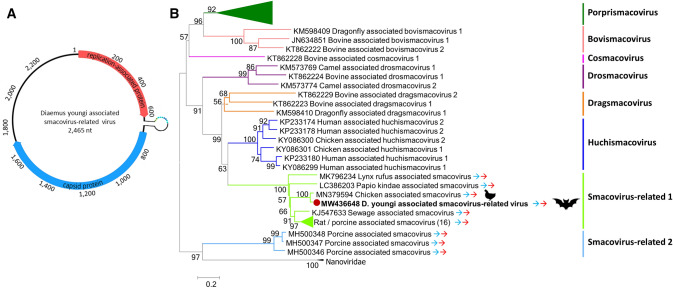
In the Neotropical region, the white-winged vampire bat (Diaemus youngi) is the rarest of the three species of vampire bats. This bat species feeds preferentially on bird blood, and there is limited information on the viruses infecting D. youngi. Hence, this study aimed to expand the knowledge about the viral diversity associated with D. youngi by sampling and pooling the lungs, liver, kidneys, heart, and intestines of all animals using high-throughput sequencing (HTS) on the Illumina MiSeq platform. A total of three complete and 10 nearly complete circular virus genomes were closely related to gemykrogvirus (Genomoviridae family), smacovirus (Smacoviridae family), and torque teno viruses (TTVs) (Anelloviridae family). In addition, three sequences of bat paramyxovirus were detected and found to be closely related to viruses reported in Pomona roundleaf bats and rodents. The present study provides a snapshot of the viral diversity associated with white-winged vampire bats and provides a baseline for comparison to viruses detected in future outbreaks.
Keywords: Diaemus youngi; High-throughput sequencing; South America; Vampire bat; Virome.
© 2022. The Author(s), under exclusive licence to Springer Science+Business Media, LLC, part of Springer Nature.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
A comparative study of rabies virus isolates from hematophagous bats in Brazil.J Wildl Dis. 2010 Oct;46(4):1335-9. doi: 10.7589/0090-3558-46.4.1335. J Wildl Dis. 2010. PMID: 20966291
-
Ultrastructure of the submandibular gland of the rare white-winged vampire bat, Diaemus youngi.Eur J Morphol. 2002 Oct;40(4):253-6. doi: 10.1076/ejom.40.4.253.16695. Eur J Morphol. 2002. PMID: 14566604
-
Genetic diversity of Bartonella spp. in vampire bats from Brazil.Transbound Emerg Dis. 2019 Nov;66(6):2329-2341. doi: 10.1111/tbed.13290. Epub 2019 Jul 24. Transbound Emerg Dis. 2019. PMID: 31287942
-
The Virome of Bats Inhabiting Brazilian Biomes: Knowledge Gaps and Biases towards Zoonotic Viruses.Microbiol Spectr. 2023 Feb 14;11(1):e0407722. doi: 10.1128/spectrum.04077-22. Epub 2023 Jan 10. Microbiol Spectr. 2023. PMID: 36625641 Free PMC article. Review.
-
Bats as Viral Reservoirs.Annu Rev Virol. 2016 Sep 29;3(1):77-99. doi: 10.1146/annurev-virology-110615-042203. Epub 2016 Aug 22. Annu Rev Virol. 2016. PMID: 27578437 Review.
Cited by
-
Corona- and Paramyxoviruses in Bats from Brazil: A Matter of Concern?Animals (Basel). 2023 Dec 26;14(1):88. doi: 10.3390/ani14010088. Animals (Basel). 2023. PMID: 38200819 Free PMC article. Review.
-
Diverse Circular DNA Viral Communities in Blood, Oral, and Fecal Samples of Captive Lemurs.Viruses. 2024 Jul 8;16(7):1099. doi: 10.3390/v16071099. Viruses. 2024. PMID: 39066262 Free PMC article.
References
-
- Jones G, Jacobs DS, Kunz TH, et al. Carpe noctem: the importance of bats as bioindicators. Endang Species Res. 2009;8:93–115. doi: 10.3354/esr00182. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
