

Pathophysiology of Lung Disease and Wound Repair in Cystic Fibrosis
- PMID: 35366275
- PMCID: PMC8830450
- DOI: 10.3390/pathophysiology28010011
Pathophysiology of Lung Disease and Wound Repair in Cystic Fibrosis
Abstract
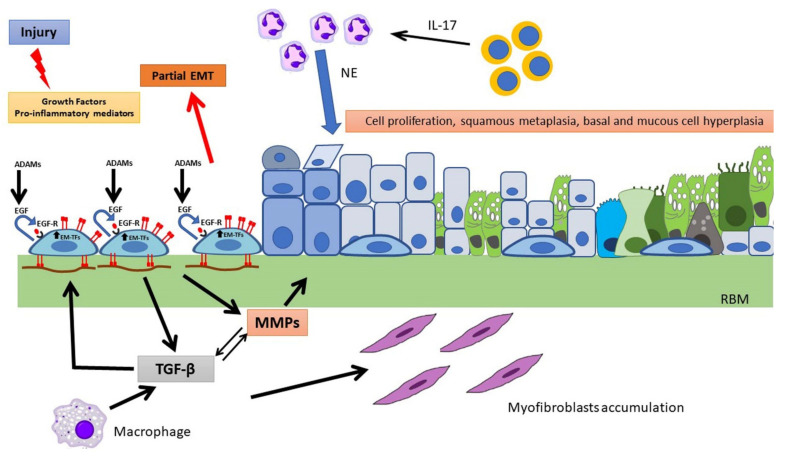
Cystic fibrosis (CF) is an autosomal recessive, life-threatening condition affecting many organs and tissues, the lung disease being the chief cause of morbidity and mortality. Mutations affecting the CF Transmembrane Conductance Regulator (CFTR) gene determine the expression of a dysfunctional protein that, in turn, triggers a pathophysiological cascade, leading to airway epithelium injury and remodeling. In vitro and in vivo studies point to a dysregulated regeneration and wound repair in CF airways, to be traced back to epithelial CFTR lack/dysfunction. Subsequent altered ion/fluid fluxes and/or signaling result in reduced cell migration and proliferation. Furthermore, the epithelial-mesenchymal transition appears to be partially triggered in CF, contributing to wound closure alteration. Finally, we pose our attention to diverse approaches to tackle this defect, discussing the therapeutic role of protease inhibitors, CFTR modulators and mesenchymal stem cells. Although the pathophysiology of wound repair in CF has been disclosed in some mechanisms, further studies are warranted to understand the cellular and molecular events in more details and to better address therapeutic interventions.
Keywords: CFTR; CFTR modulators; EGF/EGFR; airway epithelium; curcumin; cystic fibrosis; epithelial-mesenchymal transition; mesenchymal stem cells; wound healing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
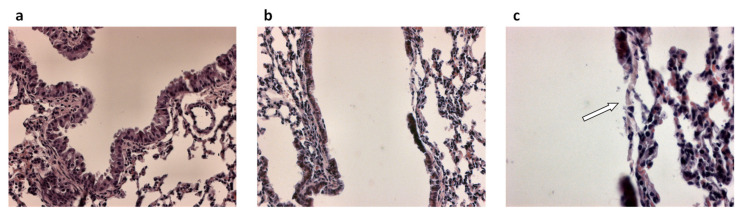
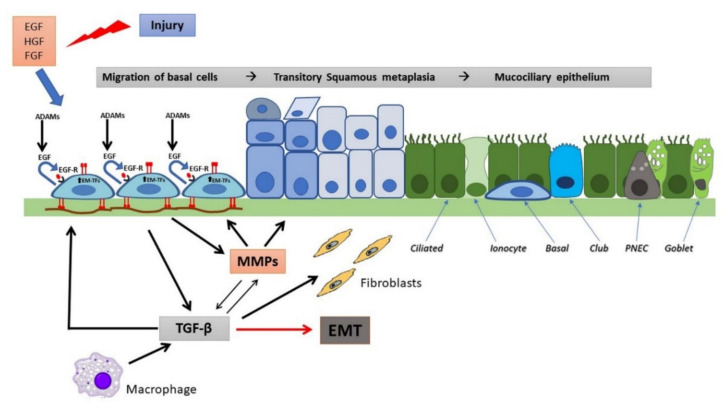
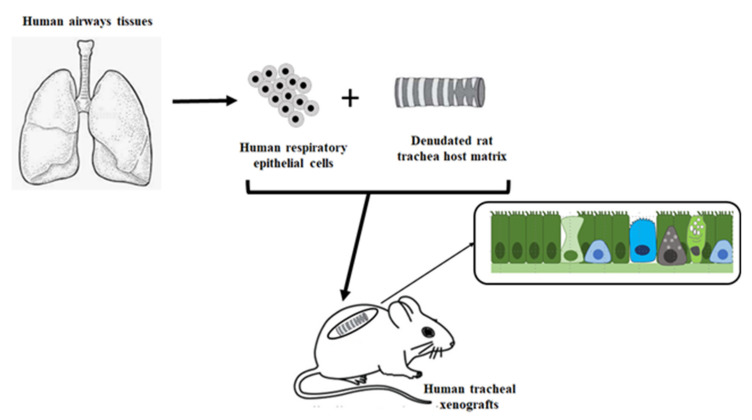
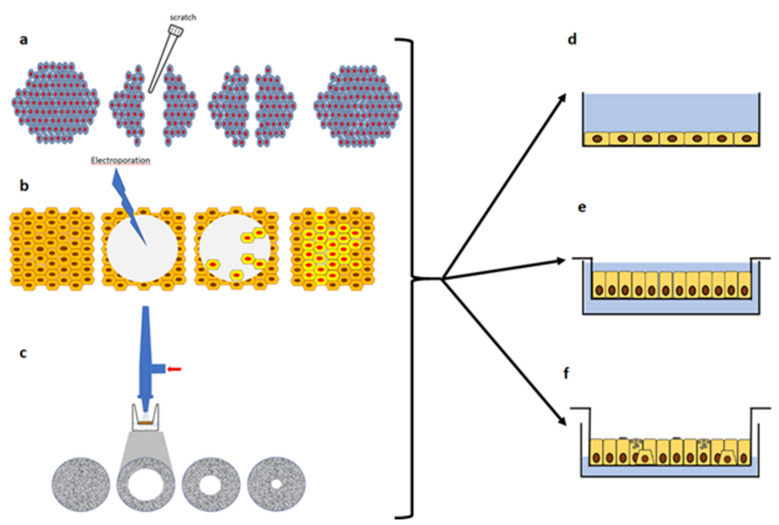
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous