

Genetic Diversity, Distribution, and Genomic Characterization of Antibiotic Resistance and Virulence of Clinical Pseudomonas aeruginosa Strains in Kenya
- PMID: 35369511
- PMCID: PMC8964364
- DOI: 10.3389/fmicb.2022.835403
Genetic Diversity, Distribution, and Genomic Characterization of Antibiotic Resistance and Virulence of Clinical Pseudomonas aeruginosa Strains in Kenya
Abstract
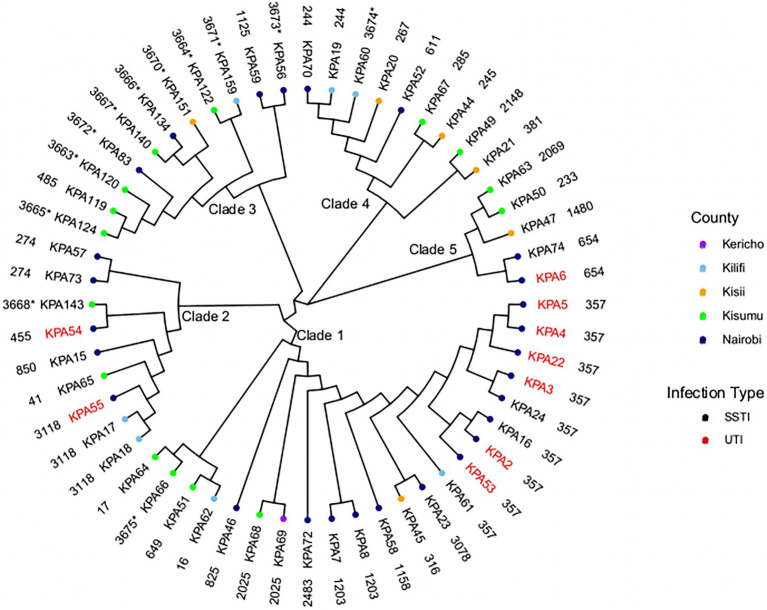
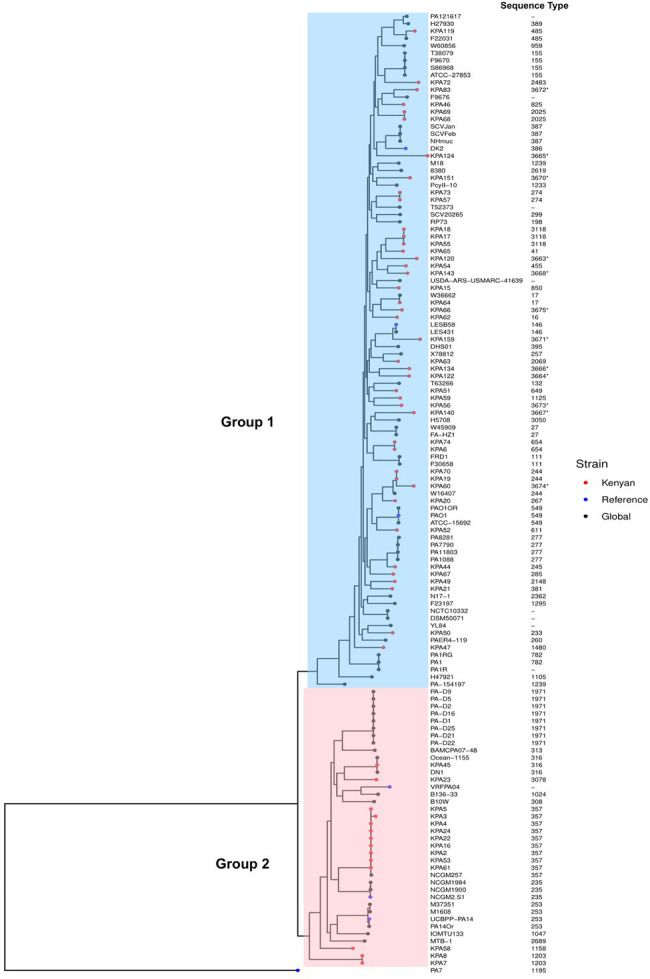
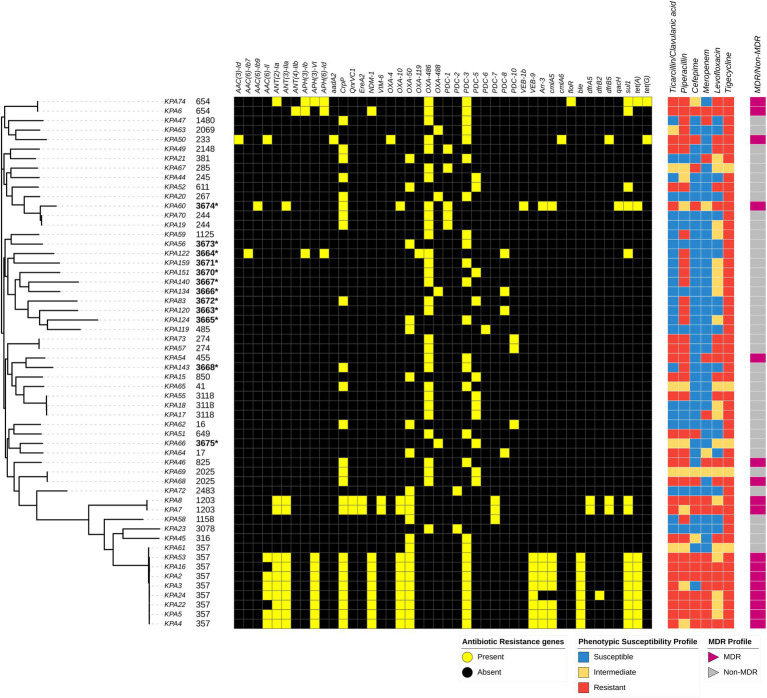
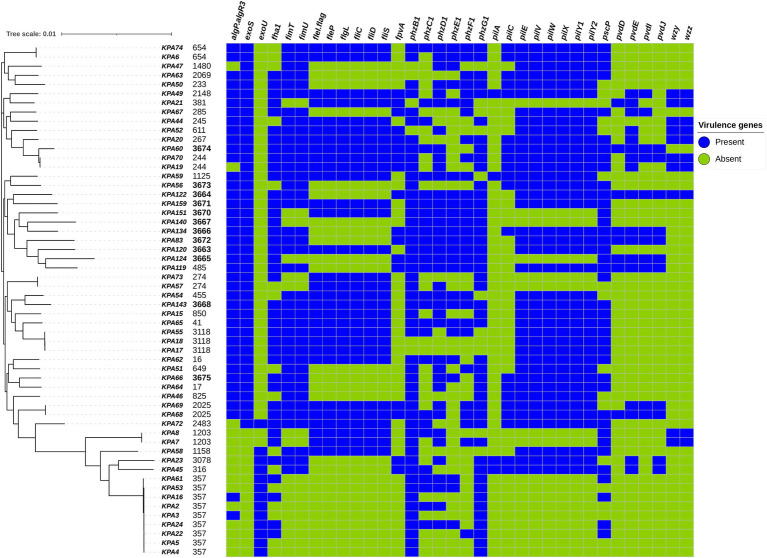
Pseudomonas aeruginosa is a leading cause of nosocomial infections worldwide. It can produce a range of debilitating infections, have a propensity for developing antimicrobial resistance, and present with a variety of potent virulence factors. This study investigated the sequence types (ST), phenotypic antimicrobial susceptibility profiles, and resistance and virulence genes among clinical isolates from urinary tract and skin and soft tissue infections. Fifty-six P. aeruginosa clinical isolates were obtained from six medical centers across five counties in Kenya between 2015 and 2020. Whole-genome sequencing (WGS) was performed to conduct genomic characterization, sequence typing, and phylogenetic analysis of the isolates. Results showed the presence of globally distributed high-risk clones (ST244 and ST357), local high-risk clones (ST2025, ST455, and ST233), and a novel multidrug-resistant (MDR) clone carrying virulence genes (ST3674). Furthermore, 31% of the study isolates were found to be MDR with phenotypic resistance to a variety of antibiotics, including piperacillin (79%), ticarcillin-clavulanic acid (57%), meropenem (34%), levofloxacin (70%), and cefepime (32%). Several resistance genes were identified, including carbapenemases VIM-6 (ST1203) and NDM-1 (ST357), fluoroquinolone genes, crpP, and qnrVCi, while 14 and 22 different chromosomal mutations were detected in the gyrA and parC genes, respectively. All isolates contained at least three virulence genes. Among the virulence genes identified, phzB1 was the most abundant (50/56, 89%). About 21% (12/56) of the isolates had the exoU+/exoS- genotype, while 73% (41/56) of the isolates had the exoS+/exoU- genotype. This study also discovered 12 novel lineages of P. aeruginosa, of which one (ST3674) demonstrated both extensive antimicrobial resistance and the highest number of virulence genes (236/242, 98%). Although most high-risk clones were detected in Nairobi County, high-risk and clones of interest were found throughout the country, indicating the local spread of global epidemic clones and the emergence of new strains. Thus, this study illustrates the urgent need for coordinated local, regional, and international antimicrobial resistance surveillance efforts.
Keywords: Kenya; Pseudomonas aeruginosa; antimicrobial resistance; sequence types; virulence.
Copyright © 2022 Kiyaga, Kyany'a, Muraya, Smith, Mills, Kibet, Mboowa and Musila.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Antimicrobial Resistance and Type III Secretion System Virulotypes of Pseudomonas aeruginosa Isolates from Dogs and Cats in Primary Veterinary Hospitals in Japan: Identification of the International High-Risk Clone Sequence Type 235.Microbiol Spectr. 2021 Oct 31;9(2):e0040821. doi: 10.1128/Spectrum.00408-21. Epub 2021 Sep 29. Microbiol Spectr. 2021. PMID: 34585944 Free PMC article.
-
Antimicrobial Activity of Ceftolozane-Tazobactam, Ceftazidime-Avibactam, and Cefiderocol against Multidrug-Resistant Pseudomonas aeruginosa Recovered at a German University Hospital.Microbiol Spectr. 2022 Oct 26;10(5):e0169722. doi: 10.1128/spectrum.01697-22. Epub 2022 Oct 3. Microbiol Spectr. 2022. PMID: 36190424 Free PMC article.
-
Molecular Characterization of Pseudomonas aeruginosa Clinical Isolates Through Whole-Genome Sequencing: A Comprehensive Analysis of Biotypes, Sequence Types, and Antimicrobial and Virulence Genes.Cureus. 2024 Oct 9;16(10):e71118. doi: 10.7759/cureus.71118. eCollection 2024 Oct. Cureus. 2024. PMID: 39525128 Free PMC article.
-
Pseudomonas aeruginosa epidemic high-risk clones and their association with horizontally-acquired β-lactamases: 2020 update.Int J Antimicrob Agents. 2020 Dec;56(6):106196. doi: 10.1016/j.ijantimicag.2020.106196. Epub 2020 Oct 9. Int J Antimicrob Agents. 2020. PMID: 33045347 Review.
-
The increasing threat of Pseudomonas aeruginosa high-risk clones.Drug Resist Updat. 2015 Jul-Aug;21-22:41-59. doi: 10.1016/j.drup.2015.08.002. Epub 2015 Aug 10. Drug Resist Updat. 2015. PMID: 26304792 Review.
Cited by
-
Enhancing capacity for national genomics surveillance of antimicrobial resistance in public health laboratories in Kenya.Microb Genom. 2023 Aug;9(8):mgen001098. doi: 10.1099/mgen.0.001098. Microb Genom. 2023. PMID: 37646415 Free PMC article.
-
Carbapenemase-producing bacteria recovered from Nairobi River, Kenya surface water and from nearby anthropogenic and zoonotic sources.PLoS One. 2024 Nov 14;19(11):e0310026. doi: 10.1371/journal.pone.0310026. eCollection 2024. PLoS One. 2024. PMID: 39541397 Free PMC article.
-
Carbapenem-resistant Pseudomonas aeruginosa T3SS virulence genes and correlation between virulence and drug resistance and molecular epidemiology studies.Front Pharmacol. 2025 Jun 18;16:1591724. doi: 10.3389/fphar.2025.1591724. eCollection 2025. Front Pharmacol. 2025. PMID: 40606623 Free PMC article.
-
Antimicrobial Resistance, Genetic Lineages, and Biofilm Formation in Pseudomonas aeruginosa Isolated from Human Infections: An Emerging One Health Concern.Antibiotics (Basel). 2023 Jul 29;12(8):1248. doi: 10.3390/antibiotics12081248. Antibiotics (Basel). 2023. PMID: 37627668 Free PMC article.
-
Are Virulence and Antibiotic Resistance Genes Linked? A Comprehensive Analysis of Bacterial Chromosomes and Plasmids.Antibiotics (Basel). 2022 May 24;11(6):706. doi: 10.3390/antibiotics11060706. Antibiotics (Basel). 2022. PMID: 35740113 Free PMC article.
References
-
- Andrews S. (2010). “FastQC: A Quality Control Tool for High Throughput Sequence Data.” 2010. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (Accessed February 15, 2021).
-
- Barrio-Tofiño E. D., López-Causapé C., Cabot G., Rivera A., Benito N., Segura C., et al. . (2017). Genomics and susceptibility profiles of extensively drug-resistant Pseudomonas aeruginosa isolates from Spain. Antimicrob. Agents Chemother. 61, e01589–e01617. doi: 10.1128/AAC.01589-17, PMID: - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous