

Phylogeny-Aware Analysis of Metagenome Community Ecology Based on Matched Reference Genomes while Bypassing Taxonomy
- PMID: 35369727
- PMCID: PMC9040630
- DOI: 10.1128/msystems.00167-22
Phylogeny-Aware Analysis of Metagenome Community Ecology Based on Matched Reference Genomes while Bypassing Taxonomy
Erratum in
-
Correction for Zhu et al., "Phylogeny-Aware Analysis of Metagenome Community Ecology Based on Matched Reference Genomes while Bypassing Taxonomy".mSystems. 2024 Nov 19;9(11):e0128924. doi: 10.1128/msystems.01289-24. Epub 2024 Oct 18. mSystems. 2024. PMID: 39422474 Free PMC article. No abstract available.
Abstract
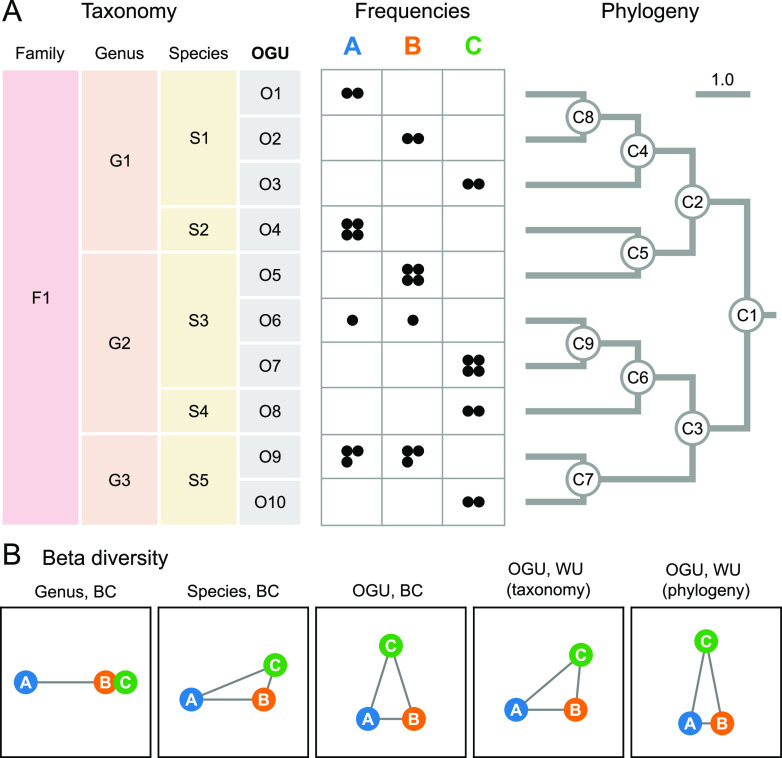
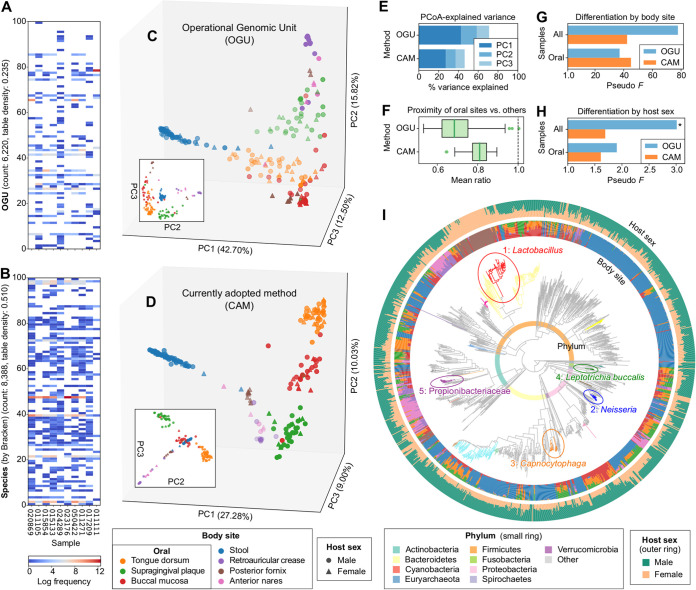
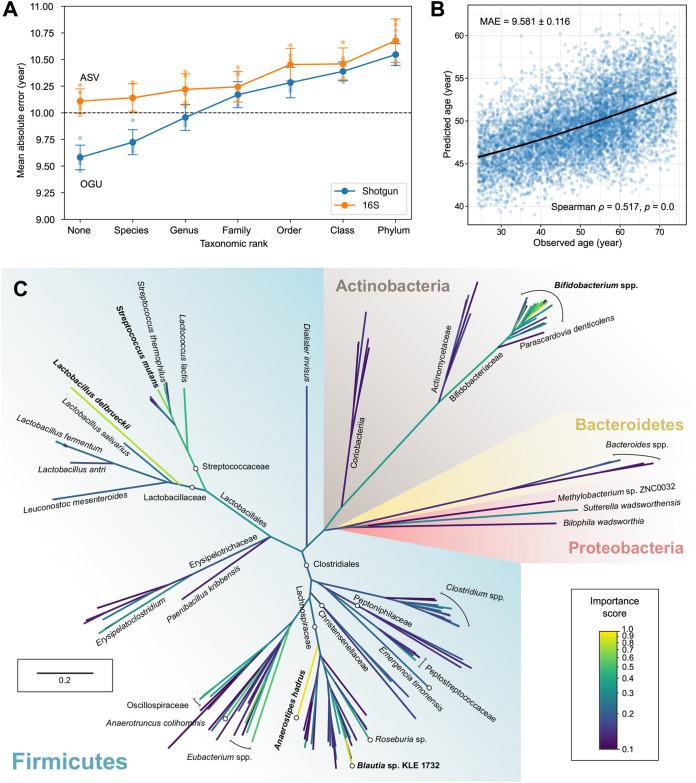
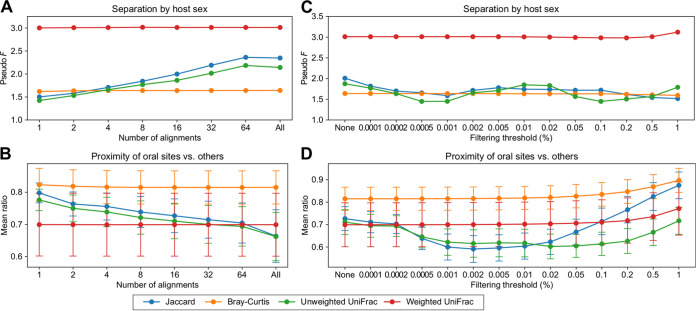
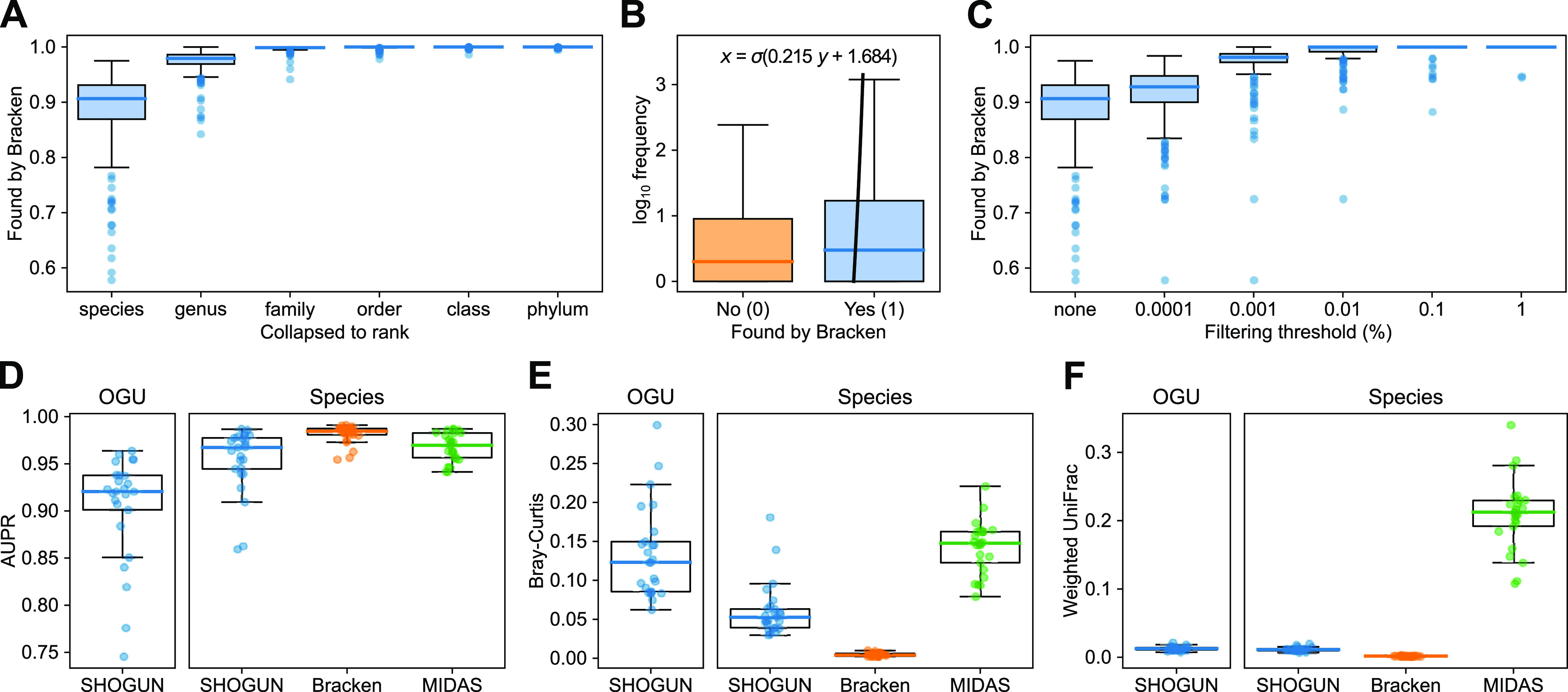
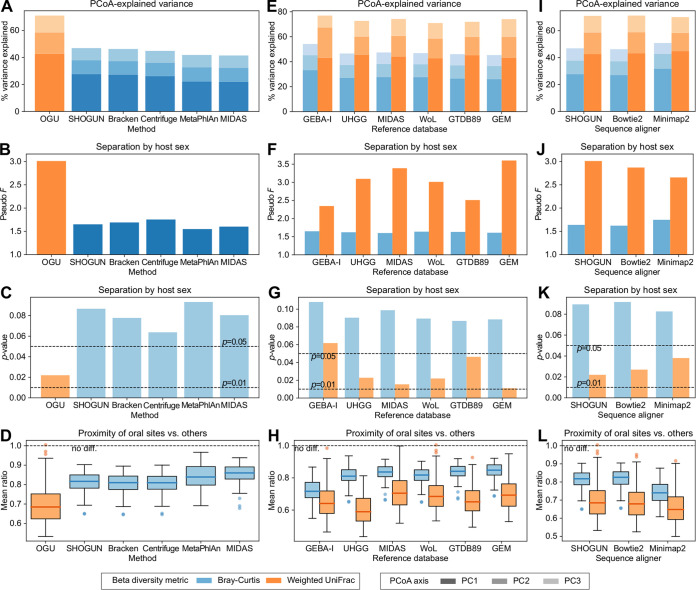
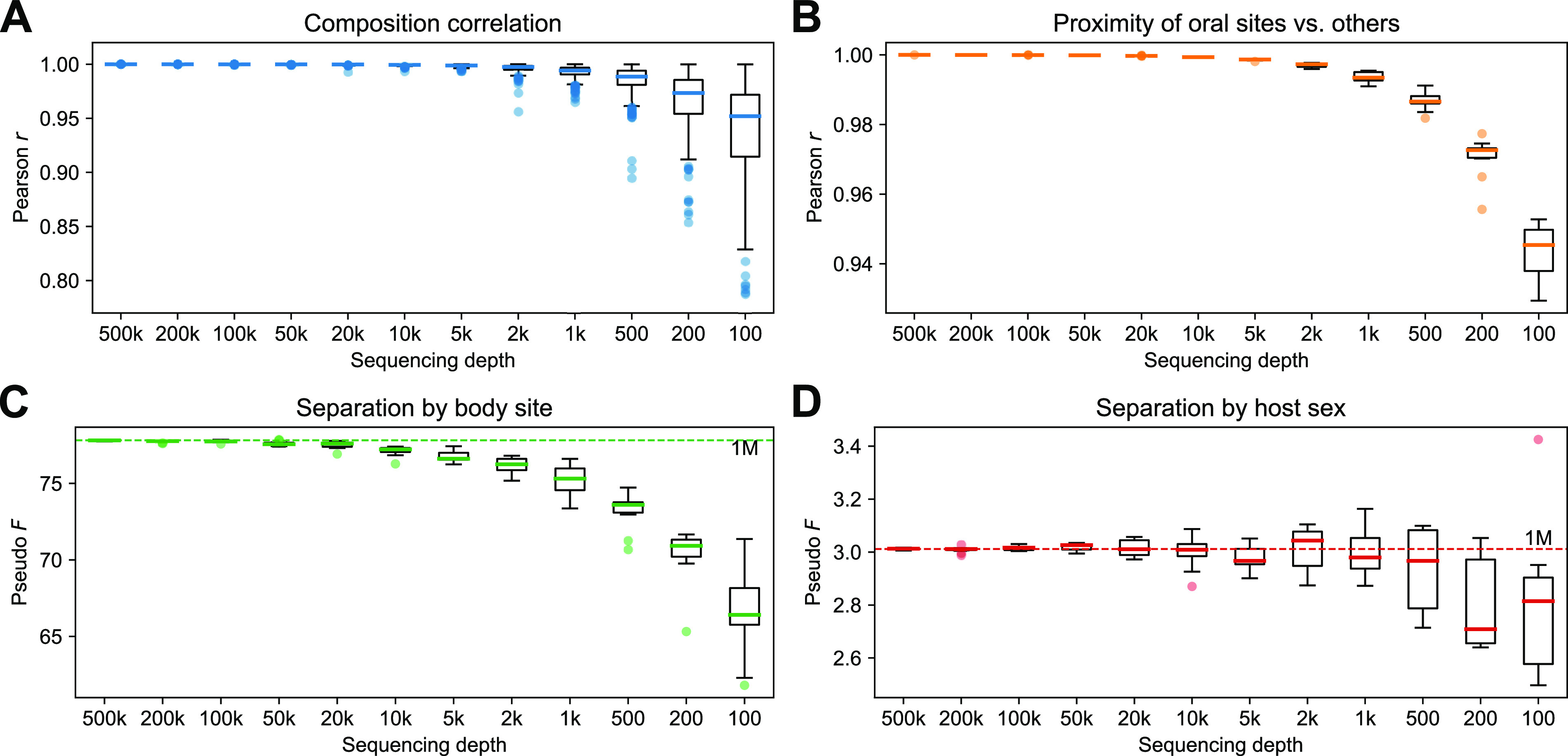
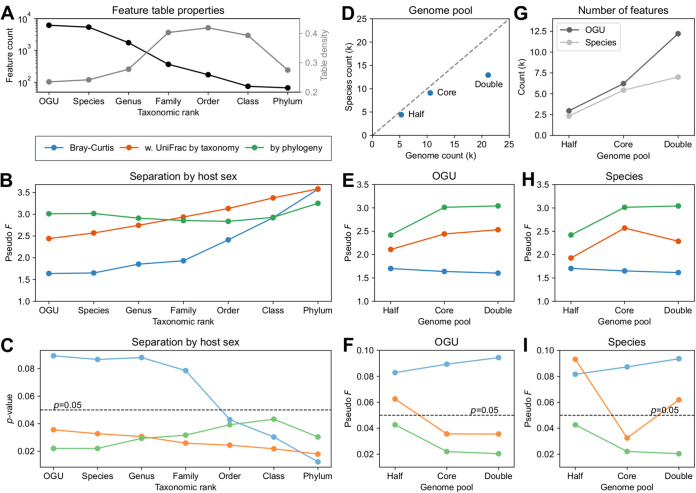
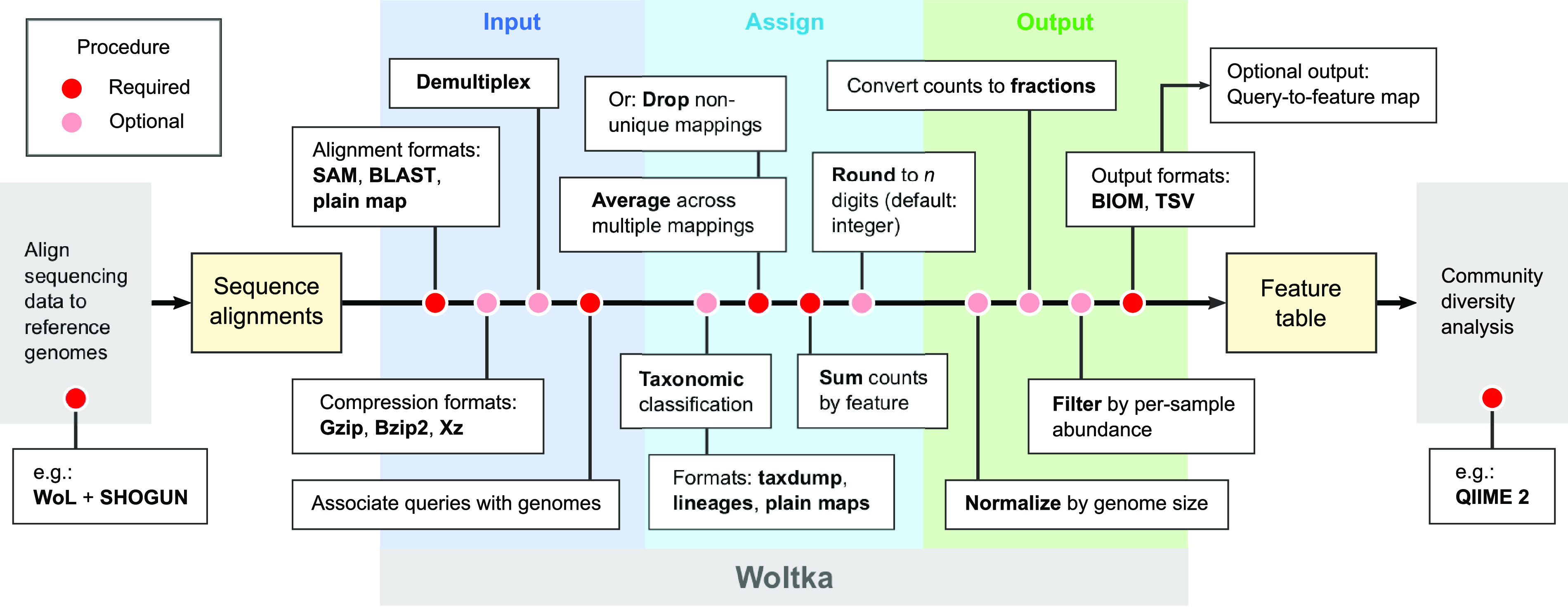
We introduce the operational genomic unit (OGU) method, a metagenome analysis strategy that directly exploits sequence alignment hits to individual reference genomes as the minimum unit for assessing the diversity of microbial communities and their relevance to environmental factors. This approach is independent of taxonomic classification, granting the possibility of maximal resolution of community composition, and organizes features into an accurate hierarchy using a phylogenomic tree. The outputs are suitable for contemporary analytical protocols for community ecology, differential abundance, and supervised learning while supporting phylogenetic methods, such as UniFrac and phylofactorization, that are seldom applied to shotgun metagenomics despite being prevalent in 16S rRNA gene amplicon studies. As demonstrated in two real-world case studies, the OGU method produces biologically meaningful patterns from microbiome data sets. Such patterns further remain detectable at very low metagenomic sequencing depths. Compared with taxonomic unit-based analyses implemented in currently adopted metagenomics tools, and the analysis of 16S rRNA gene amplicon sequence variants, this method shows superiority in informing biologically relevant insights, including stronger correlation with body environment and host sex on the Human Microbiome Project data set and more accurate prediction of human age by the gut microbiomes of Finnish individuals included in the FINRISK 2002 cohort. We provide Woltka, a bioinformatics tool to implement this method, with full integration with the QIIME 2 package and the Qiita web platform, to facilitate adoption of the OGU method in future metagenomics studies. IMPORTANCE Shotgun metagenomics is a powerful, yet computationally challenging, technique compared to 16S rRNA gene amplicon sequencing for decoding the composition and structure of microbial communities. Current analyses of metagenomic data are primarily based on taxonomic classification, which is limited in feature resolution. To solve these challenges, we introduce operational genomic units (OGUs), which are the individual reference genomes derived from sequence alignment results, without further assigning them taxonomy. The OGU method advances current read-based metagenomics in two dimensions: (i) providing maximal resolution of community composition and (ii) permitting use of phylogeny-aware tools. Our analysis of real-world data sets shows that it is advantageous over currently adopted metagenomic analysis methods and the finest-grained 16S rRNA analysis methods in predicting biological traits. We thus propose the adoption of OGUs as an effective practice in metagenomic studies.
Keywords: UniFrac; metagenomics; operational genomic unit; reference phylogeny; supervised learning; taxonomy independent.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
