

High-resolution Slide-seqV2 spatial transcriptomics enables discovery of disease-specific cell neighborhoods and pathways
- PMID: 35372810
- PMCID: PMC8971939
- DOI: 10.1016/j.isci.2022.104097
High-resolution Slide-seqV2 spatial transcriptomics enables discovery of disease-specific cell neighborhoods and pathways
Abstract
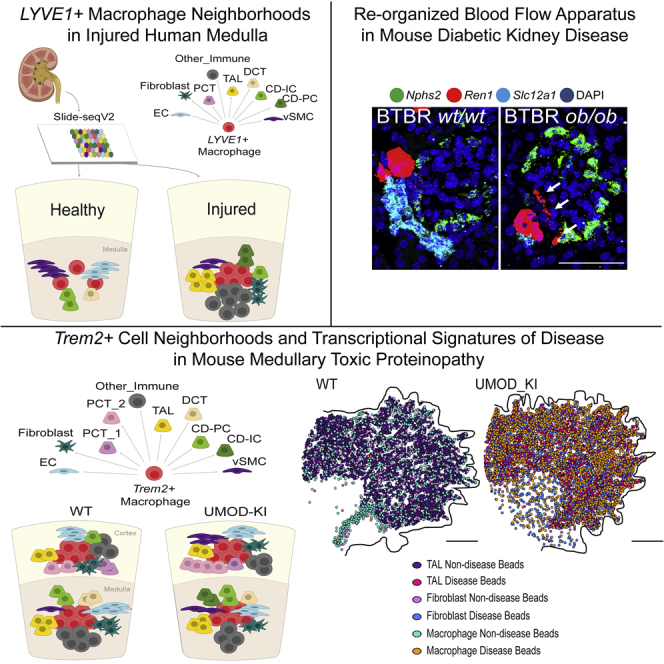
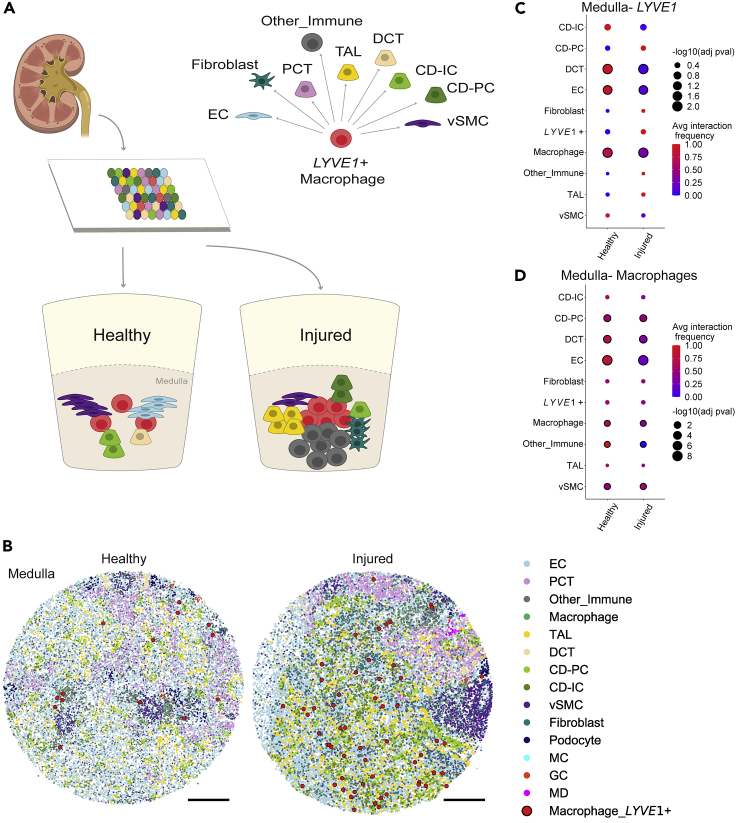
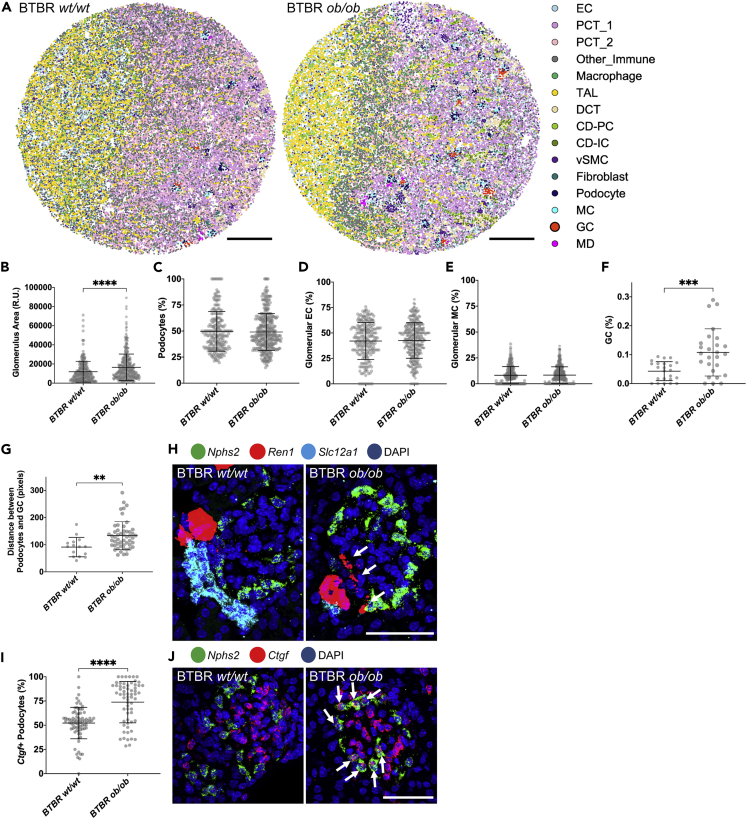
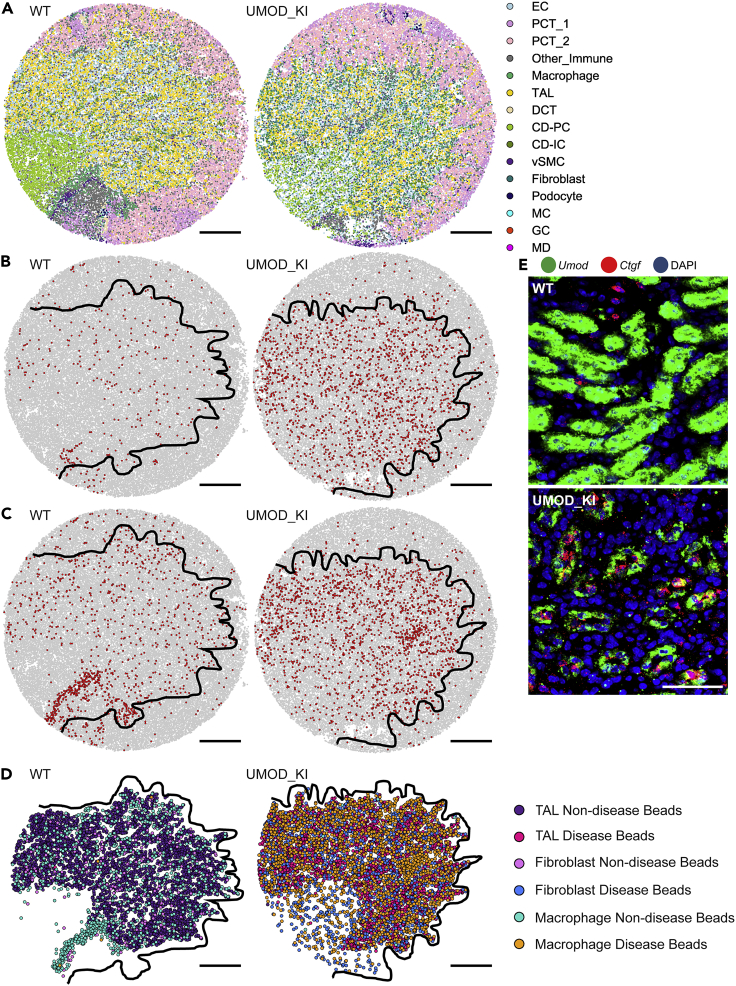
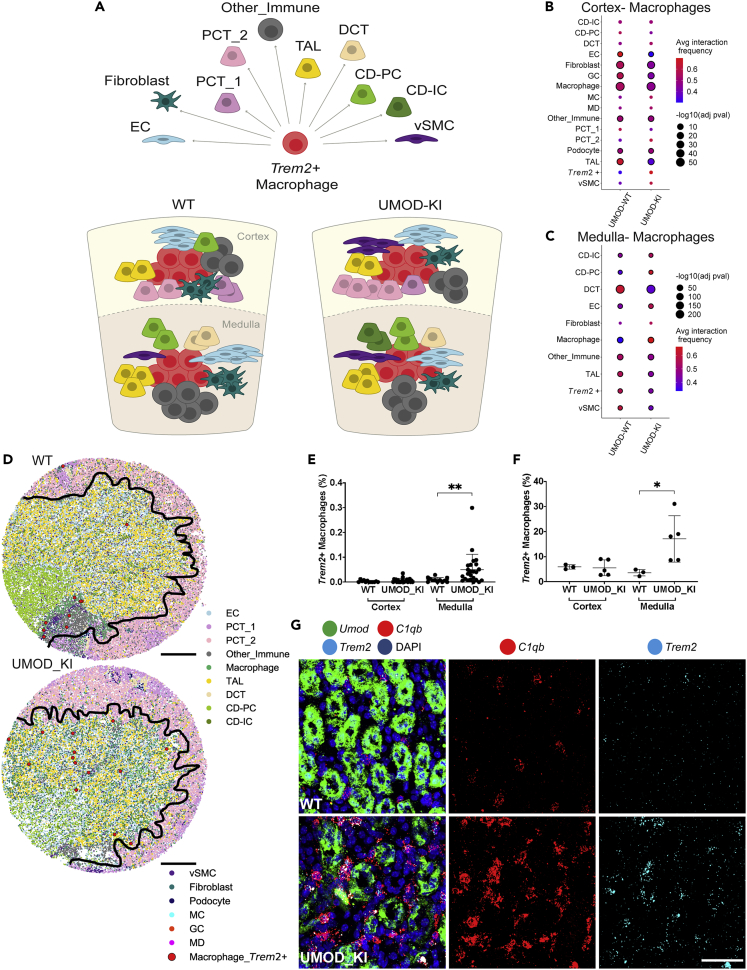
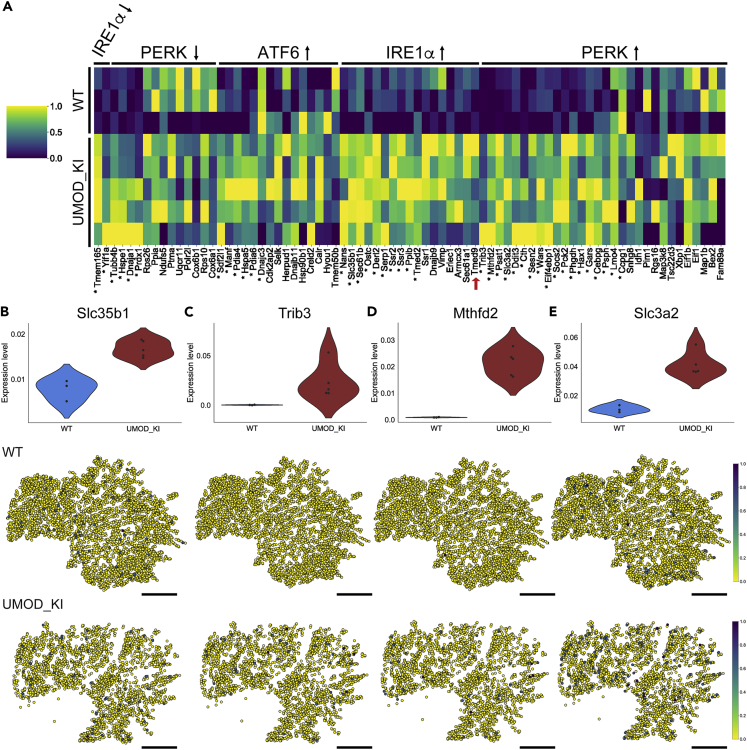
High-resolution spatial transcriptomics enables mapping of RNA expression directly from intact tissue sections; however, its utility for the elucidation of disease processes and therapeutically actionable pathways remains unexplored. We applied Slide-seqV2 to mouse and human kidneys, in healthy and distinct disease paradigms. First, we established the feasibility of Slide-seqV2 in tissue from nine distinct human kidneys, which revealed a cell neighborhood centered around a population of LYVE1+ macrophages. Second, in a mouse model of diabetic kidney disease, we detected changes in the cellular organization of the spatially restricted kidney filter and blood-flow-regulating apparatus. Third, in a mouse model of a toxic proteinopathy, we identified previously unknown, disease-specific cell neighborhoods centered around macrophages. In a spatially restricted subpopulation of epithelial cells, we discovered perturbations in 77 genes associated with the unfolded protein response. Our studies illustrate and experimentally validate the utility of Slide-seqV2 for the discovery of disease-specific cell neighborhoods.
Keywords: Cell biology; Pathophysiology; Transcriptomics.
© 2022 The Author(s).
Conflict of interest statement
AG serves as a founding advisor to Goldfinch Biopharma and to a new Atlas Ventures funded company, with respective agreements reviewed and managed by Mass General Brigham (MGB) and the Broad Institute of MIT and Harvard in accordance with their conflict of interest policies. RRS, FC, and EZM are inventors on a pending patent application related to the development of Slide-seq. EZM is an advisor to Curio Biosciences, Inc. FC is a paid consultant for Atlas Bio. KAV is currently an employee and shareholder of Q32 Bio, Inc. JLM is currently an employee and shareholder of Solid Biosciences, Inc.
Figures
References
-
- Bachmann S., Oberbäumer I. Structural and molecular dissection of the juxtaglomerular apparatus: new aspects for the role of nitric oxide. Kidney Int. Suppl. 1998;67:S29–S33. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
