

Gain of function due to increased opening probability by two KCNQ5 pore variants causing developmental and epileptic encephalopathy
- PMID: 35377796
- PMCID: PMC9169635
- DOI: 10.1073/pnas.2116887119
Gain of function due to increased opening probability by two KCNQ5 pore variants causing developmental and epileptic encephalopathy
Abstract
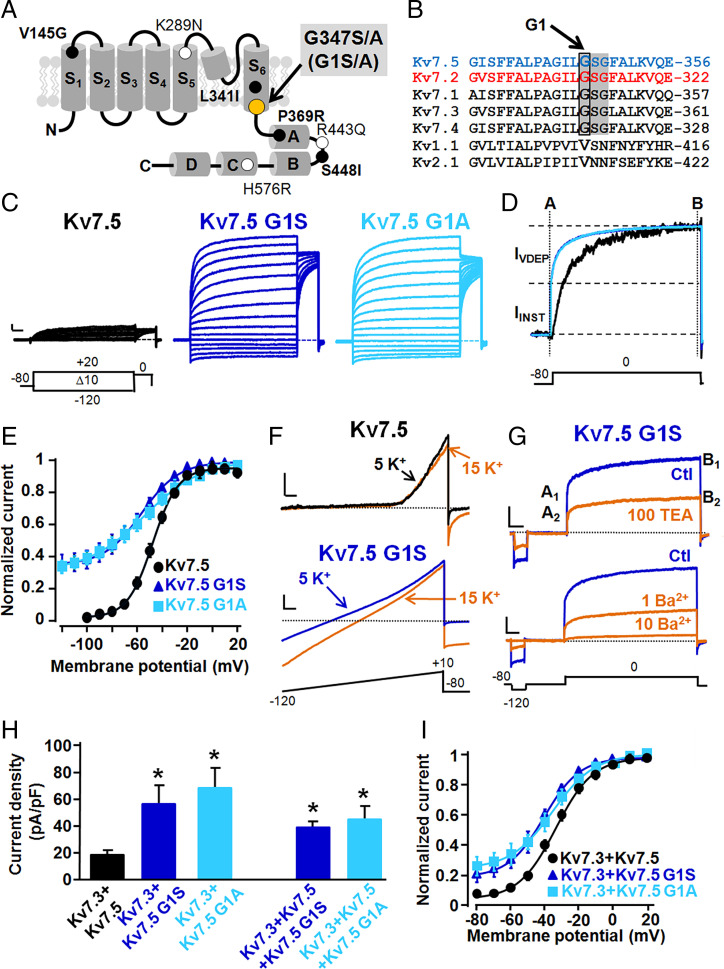
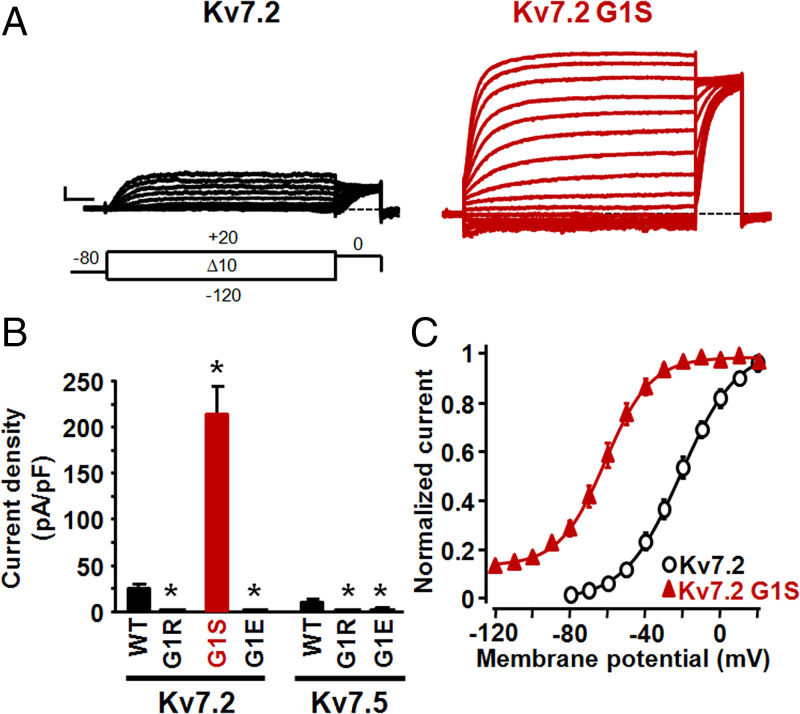
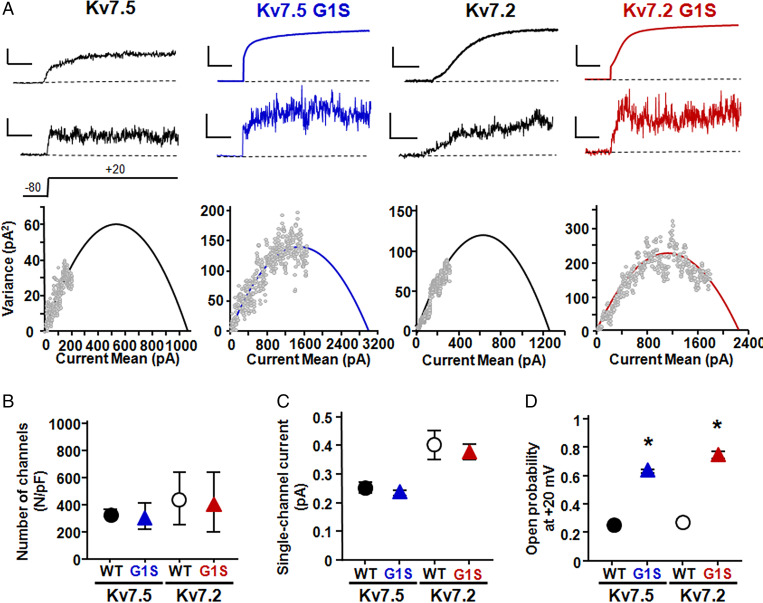
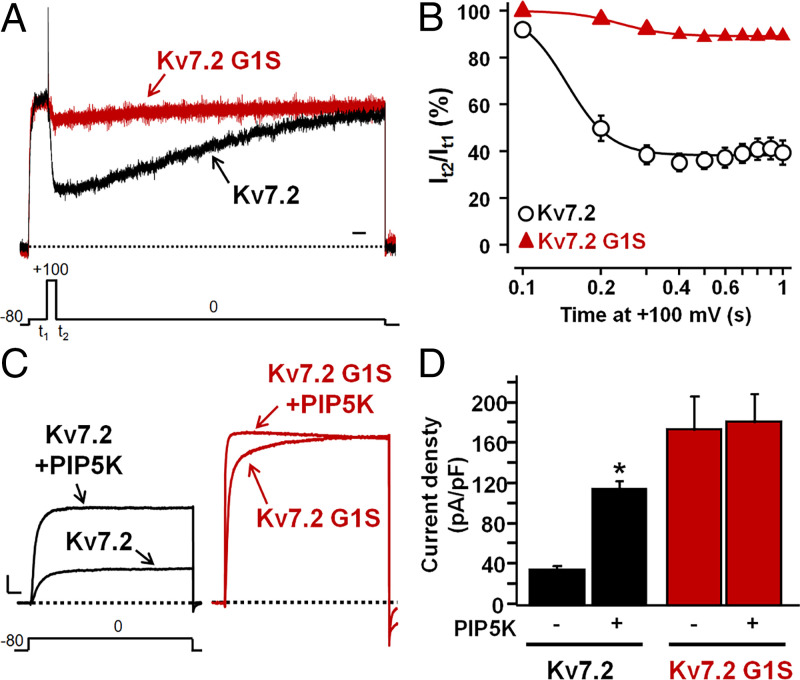
Developmental and epileptic encephalopathies (DEEs) are neurodevelopmental diseases characterized by refractory epilepsy, distinct electroencephalographic and neuroradiological features, and various degrees of developmental delay. Mutations in KCNQ2, KCNQ3, and, more rarely, KCNQ5 genes encoding voltage-gated potassium channel subunits variably contributing to excitability control of specific neuronal populations at distinct developmental stages have been associated to DEEs. In the present work, the clinical features of two DEE patients carrying de novo KCNQ5 variants affecting the same residue in the pore region of the Kv7.5 subunit (G347S/A) are described. The in vitro functional properties of channels incorporating these variants were investigated with electrophysiological and biochemical techniques to highlight pathophysiological disease mechanisms. Currents carried by Kv7.5 G347 S/A channels displayed: 1) large (>10 times) increases in maximal current density, 2) the occurrence of a voltage-independent component, 3) slower deactivation kinetics, and 4) hyperpolarization shift in activation. All these functional features are consistent with a gain-of-function (GoF) pathogenetic mechanism. Similar functional changes were also observed when the same variants were introduced at the corresponding position in Kv7.2 subunits. Nonstationary noise analysis revealed that GoF effects observed for both Kv7.2 and Kv7.5 variants were mainly attributable to an increase in single-channel open probability, without changes in membrane abundance or single-channel conductance. The mutation-induced increase in channel opening probability was insensitive to manipulation of membrane levels of the critical Kv7 channel regulator PIP2. These results reveal a pathophysiological mechanism for KCNQ5-related DEEs, which might be exploited to implement personalized treatments.
Keywords: developmental and epileptic encephalopathies; genotype–phenotype correlations; potassium channels.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
-
- Kearney H., Byrne S., Cavalleri G. L., Delanty N., Tackling epilepsy with high-definition precision medicine: A review. JAMA Neurol. 76, 1109–1116 (2019). - PubMed
-
- Jones F., Gamper N., Gao H., “Kv7 channels and excitability disorders” in Handbook of Experimental Pharmacology, Gamper N., Wang K., Eds. (Springer, 2021), pp. 185–230. - PubMed
-
- Lerche C., et al. , Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J. Biol. Chem. 275, 22395–22400 (2000). - PubMed
-
- Schroeder B. C., Hechenberger M., Weinreich F., Kubisch C., Jentsch T. J., KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J. Biol. Chem. 275, 24089–24095 (2000). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
