

Neurodevelopmental disorder with microcephaly, hypotonia, and variable brain anomalies in a consanguineous Iranian family is associated with a homozygous start loss variant in the PRUNE1 gene
- PMID: 35379233
- PMCID: PMC8981834
- DOI: 10.1186/s12920-022-01228-6
Neurodevelopmental disorder with microcephaly, hypotonia, and variable brain anomalies in a consanguineous Iranian family is associated with a homozygous start loss variant in the PRUNE1 gene
Abstract
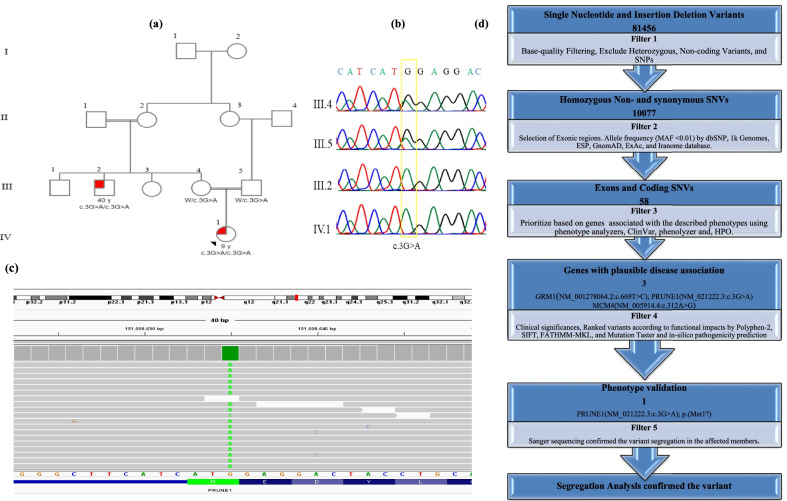
Background: Homozygous or compound heterozygous PRUNE1 mutations cause a neurodevelopmental disorder with microcephaly, hypotonia, and variable brain malformations (NMIHBA) (OMIM #617481). The PRUNE1 gene encodes a member of the phosphoesterase (DHH) protein superfamily that is involved in the regulation of cell migration. To date, most of the described mutations in the PRUNE1 gene are clustered in DHH domain.
Methods: We subjected 4 members (two affected and two healthy) of a consanguineous Iranian family in the study. The proband underwent whole-exome sequencing and a start loss identified variant was confirmed by Sanger sequencing. Co-segregation of the detected variant with the disease in family was confirmed.
Results: By whole-exome sequencing, we identified the a start loss variant, NM_021222.3:c.3G>A; p.(Met1?), in the PRUNE1 in two patients of a consanguineous Iranian family with spastic quadriplegic cerebral palsy (CP), hypotonia, developmental regression, and cerebellar atrophy. Sanger sequencing confirmed the segregation of the variant with the disease in the family. Protein structure analysis also revealed that the variant probably leads to the deletion of DHH (Asp-His-His) domain, the active site of the protein, and loss of PRUNE1 function.
Conclusion: We identified a start loss variant, NM_021222.3:c.3G>A; p.(Met1?) in the PRUNE1 gene in two affected members as a possible cause of NMIHBA in an Iranian family. We believe that the study adds a new pathogenic variant in spectrum of mutations in the PRUNE1 gene as a cause of PRUNE1-related syndrome.
Keywords: NMIHBA; Neurodevelopmental disorder; PRUNE1; Whole-exome sequencing.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
