

Clinical implementation of RNA sequencing for Mendelian disease diagnostics
- PMID: 35379322
- PMCID: PMC8981716
- DOI: 10.1186/s13073-022-01019-9
Clinical implementation of RNA sequencing for Mendelian disease diagnostics
Abstract
Background: Lack of functional evidence hampers variant interpretation, leaving a large proportion of individuals with a suspected Mendelian disorder without genetic diagnosis after whole genome or whole exome sequencing (WES). Research studies advocate to further sequence transcriptomes to directly and systematically probe gene expression defects. However, collection of additional biopsies and establishment of lab workflows, analytical pipelines, and defined concepts in clinical interpretation of aberrant gene expression are still needed for adopting RNA sequencing (RNA-seq) in routine diagnostics.
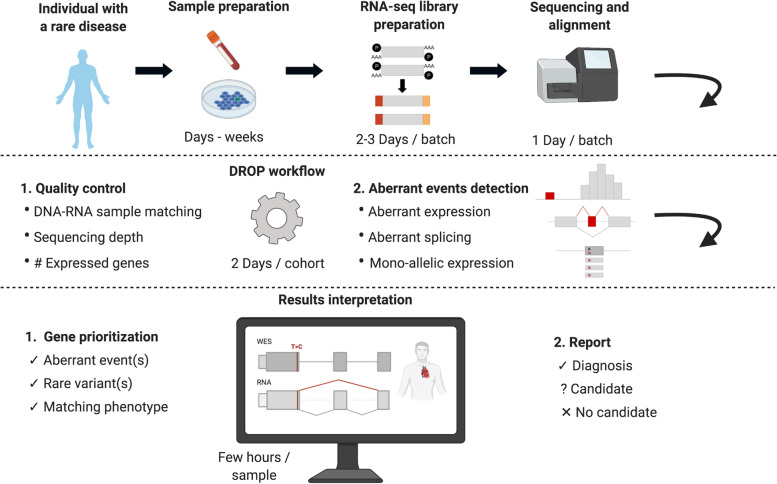
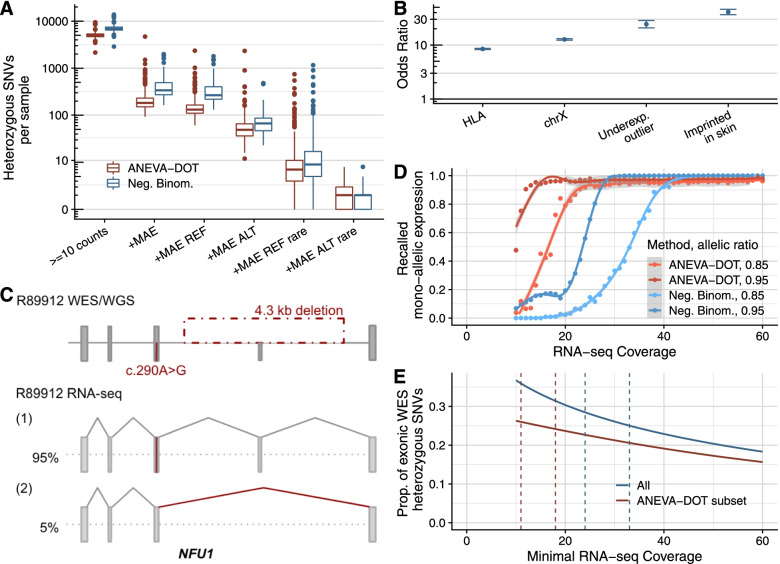
Methods: We implemented an automated RNA-seq protocol and a computational workflow with which we analyzed skin fibroblasts of 303 individuals with a suspected mitochondrial disease that previously underwent WES. We also assessed through simulations how aberrant expression and mono-allelic expression tests depend on RNA-seq coverage.
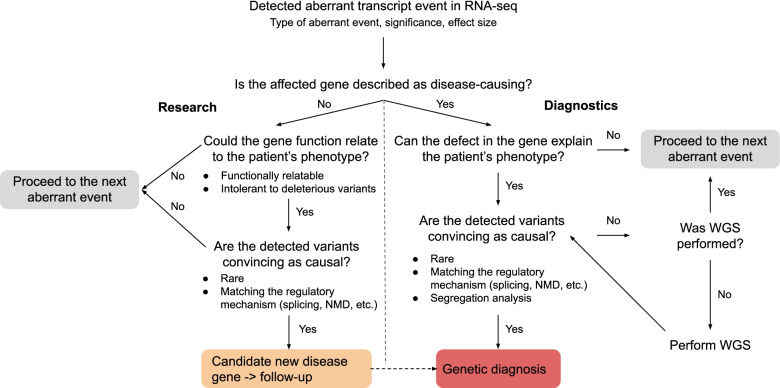
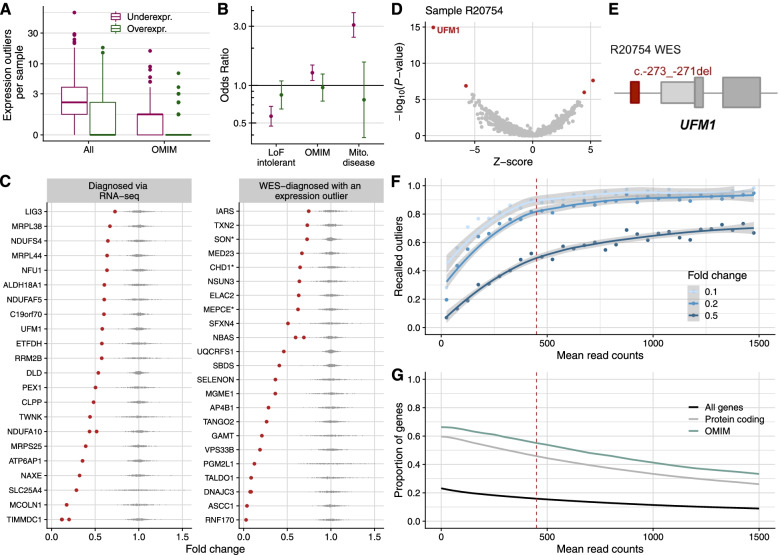
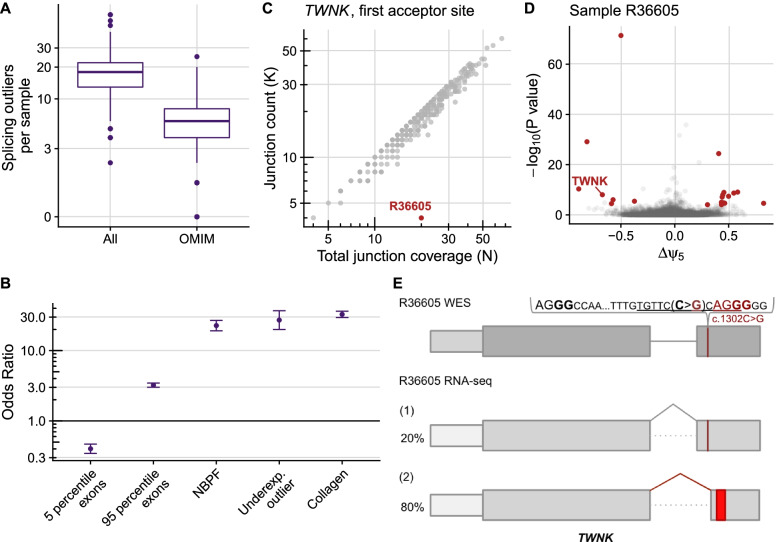
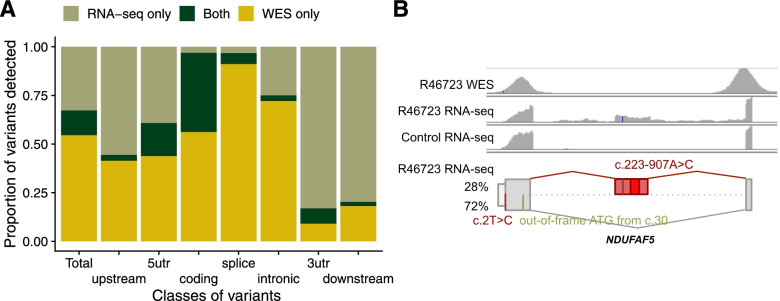
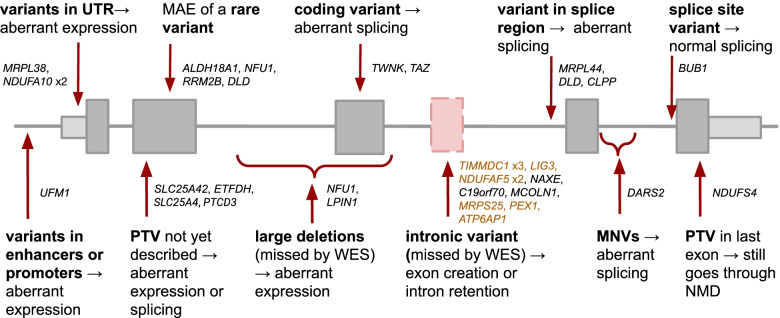
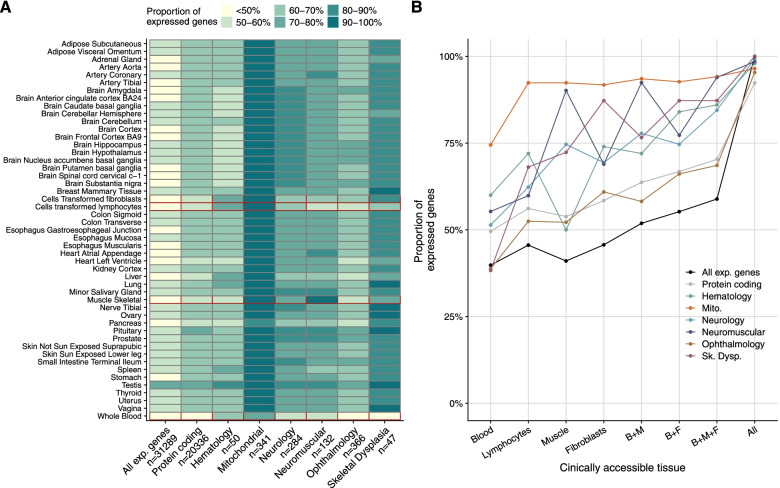
Results: We detected on average 12,500 genes per sample including around 60% of all disease genes-a coverage substantially higher than with whole blood, supporting the use of skin biopsies. We prioritized genes demonstrating aberrant expression, aberrant splicing, or mono-allelic expression. The pipeline required less than 1 week from sample preparation to result reporting and provided a median of eight disease-associated genes per patient for inspection. A genetic diagnosis was established for 16% of the 205 WES-inconclusive cases. Detection of aberrant expression was a major contributor to diagnosis including instances of 50% reduction, which, together with mono-allelic expression, allowed for the diagnosis of dominant disorders caused by haploinsufficiency. Moreover, calling aberrant splicing and variants from RNA-seq data enabled detecting and validating splice-disrupting variants, of which the majority fell outside WES-covered regions.
Conclusion: Together, these results show that streamlined experimental and computational processes can accelerate the implementation of RNA-seq in routine diagnostics.
Keywords: Genetic diagnostics; Mendelian diseases; RNA-seq.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- EURORDIS. Rare Diseases: Understanding this Public Health Priority. Rare Dis. 2005;1–14.
-
- Wright CF, FitzPatrick DR, Firth HV. Paediatric genomics: diagnosing rare disease in children. Nat Rev Genet. 2018;19(5):253–68. - PubMed
-
- Koch J, Mayr JA, Alhaddad B, Rauscher C, Bierau J, Kovacs-Nagy R, et al. CAD mutations and uridine-responsive epileptic encephalopathy. Brain. 2017;140(2):279–286. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
