

Cannabis suppresses antitumor immunity by inhibiting JAK/STAT signaling in T cells through CNR2
- PMID: 35383142
- PMCID: PMC8983672
- DOI: 10.1038/s41392-022-00918-y
Cannabis suppresses antitumor immunity by inhibiting JAK/STAT signaling in T cells through CNR2
Abstract
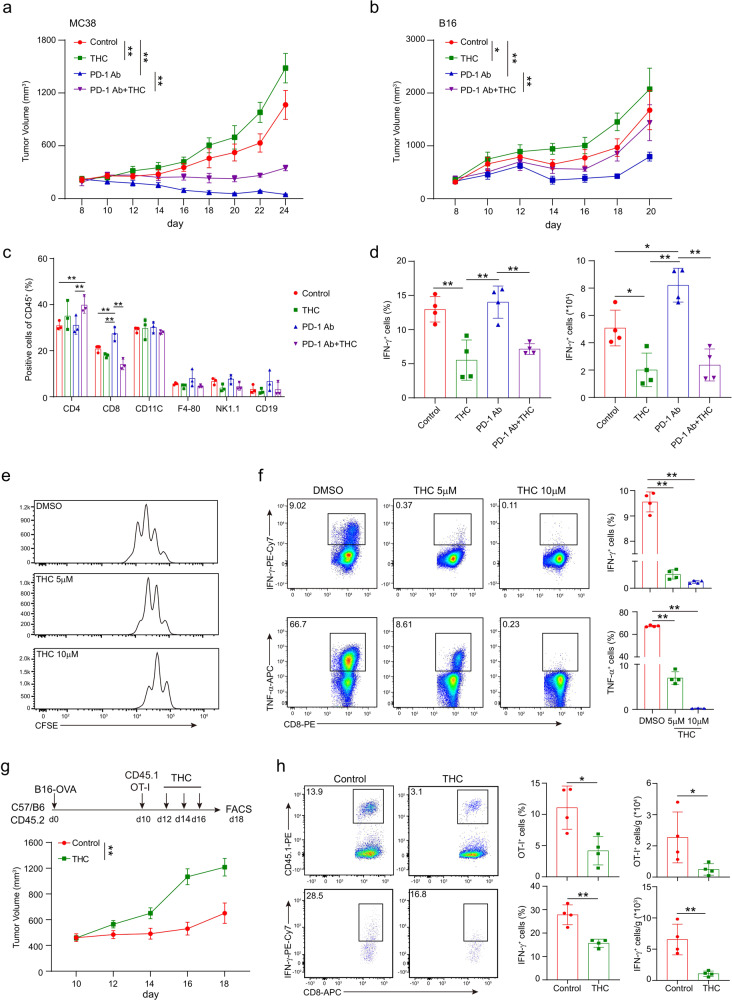
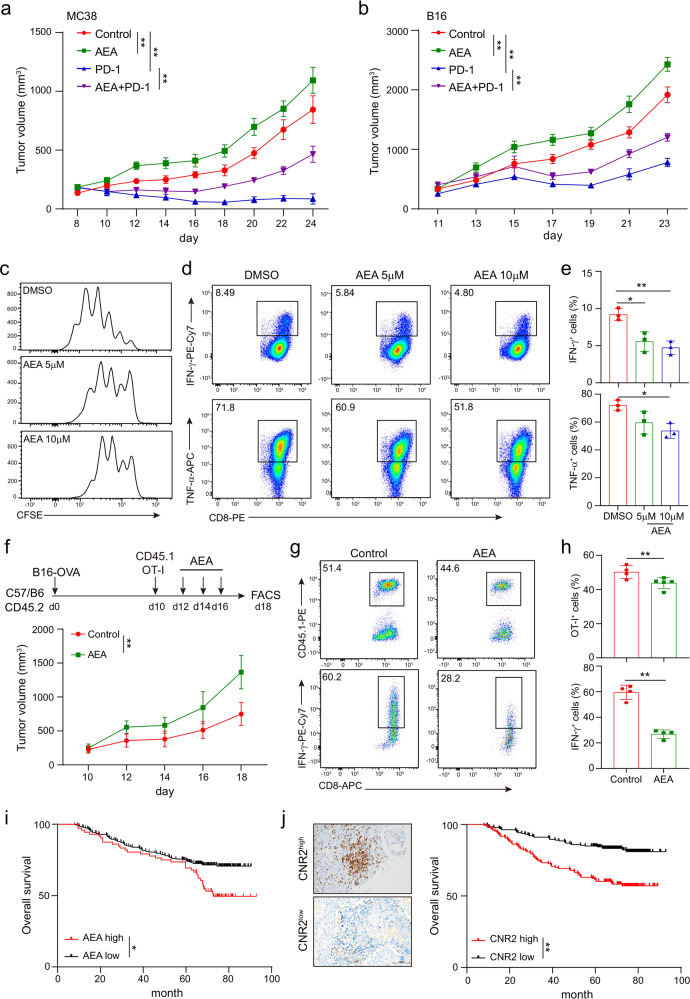
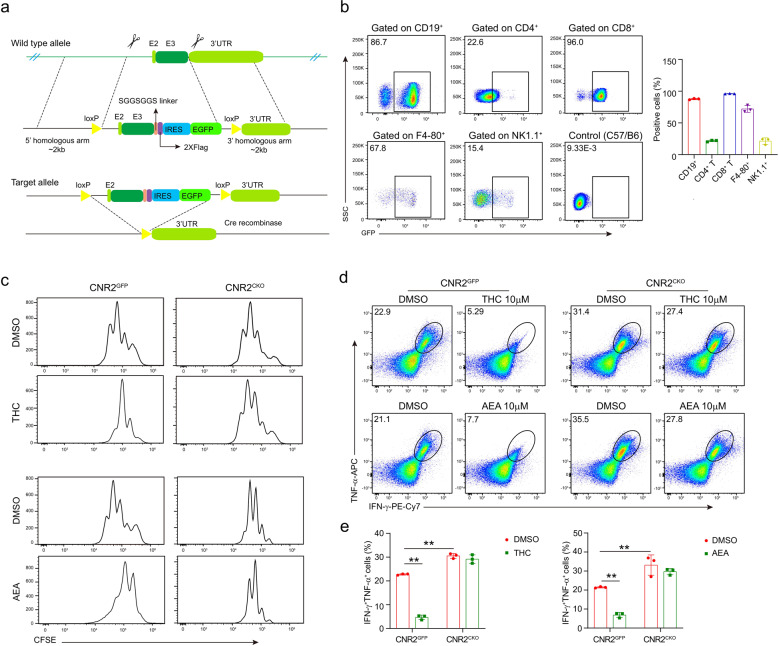
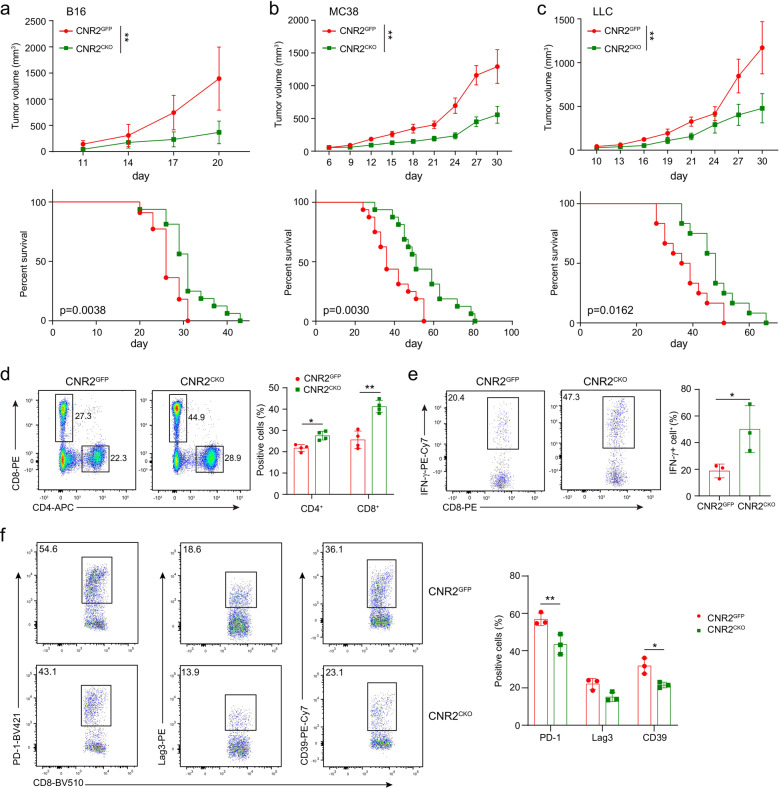
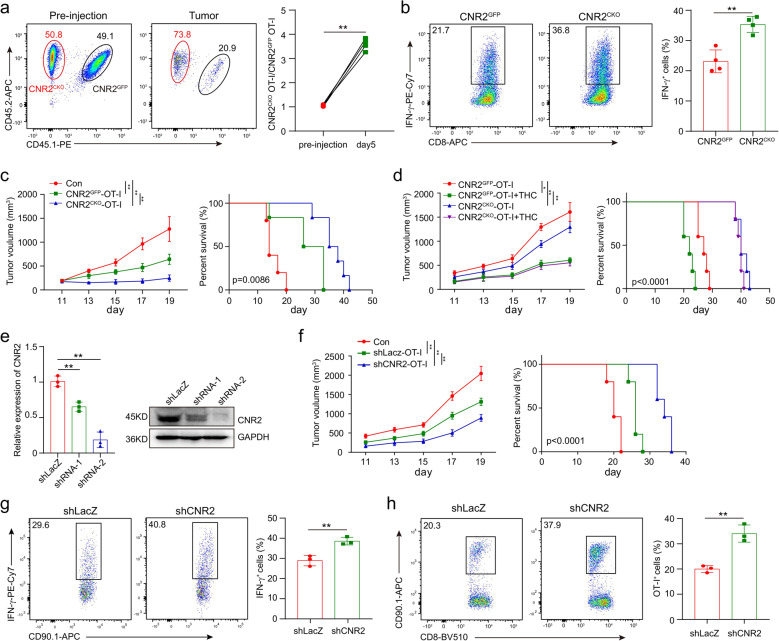
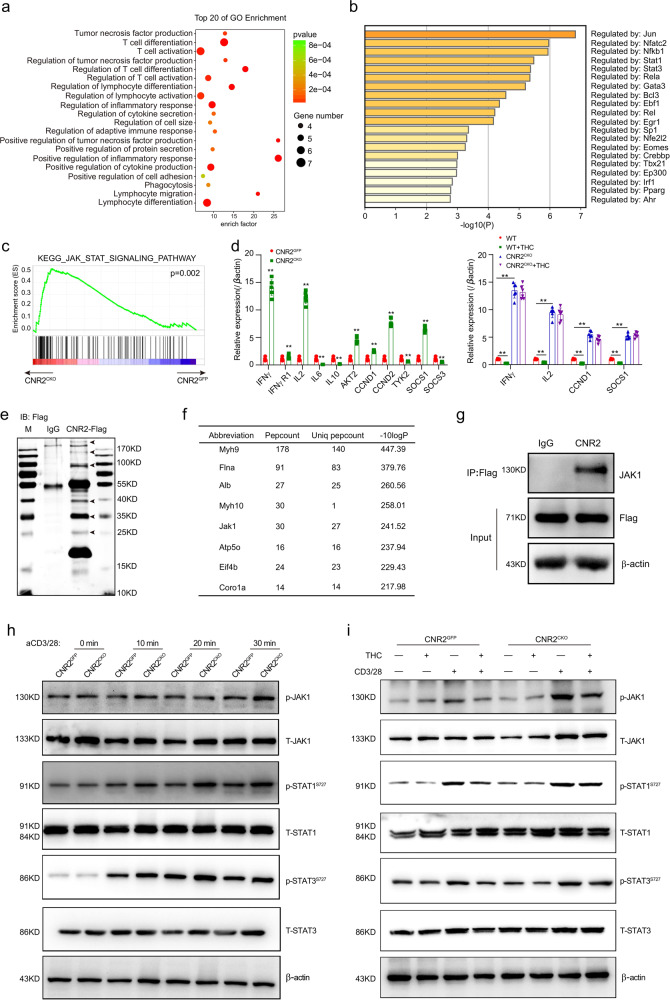
The combination of immune checkpoint blockade (ICB) with chemotherapy significantly improves clinical benefit of cancer treatment. Since chemotherapy is often associated with adverse events, concomitant treatment with drugs managing side effects of chemotherapy is frequently used in the combination therapy. However, whether these ancillary drugs could impede immunotherapy remains unknown. Here, we showed that ∆9-tetrahydrocannabinol (THC), the key ingredient of drugs approved for the treatment of chemotherapy-caused nausea, reduced the therapeutic effect of PD-1 blockade. The endogenous cannabinoid anandamide (AEA) also impeded antitumor immunity, indicating an immunosuppressive role of the endogenous cannabinoid system (ECS). Consistently, high levels of AEA in the sera were associated with poor overall survival in cancer patients. We further found that cannabinoids impaired the function of tumor-specific T cells through CNR2. Using a knock-in mouse model expressing a FLAG-tagged Cnr2 gene, we discovered that CNR2 binds to JAK1 and inhibits the downstream STAT signaling in T cells. Taken together, our results unveiled a novel mechanism of the ECS-mediated suppression on T-cell immunity against cancer, and suggest that cannabis and cannabinoid drugs should be avoided during immunotherapy.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
