

High-throughput translational profiling with riboPLATE-seq
- PMID: 35383235
- PMCID: PMC8983706
- DOI: 10.1038/s41598-022-09638-3
High-throughput translational profiling with riboPLATE-seq
Abstract
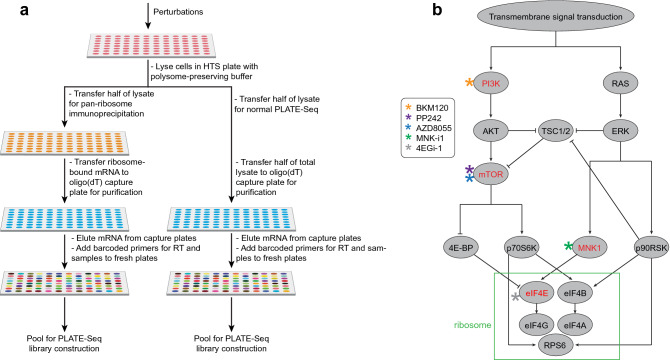
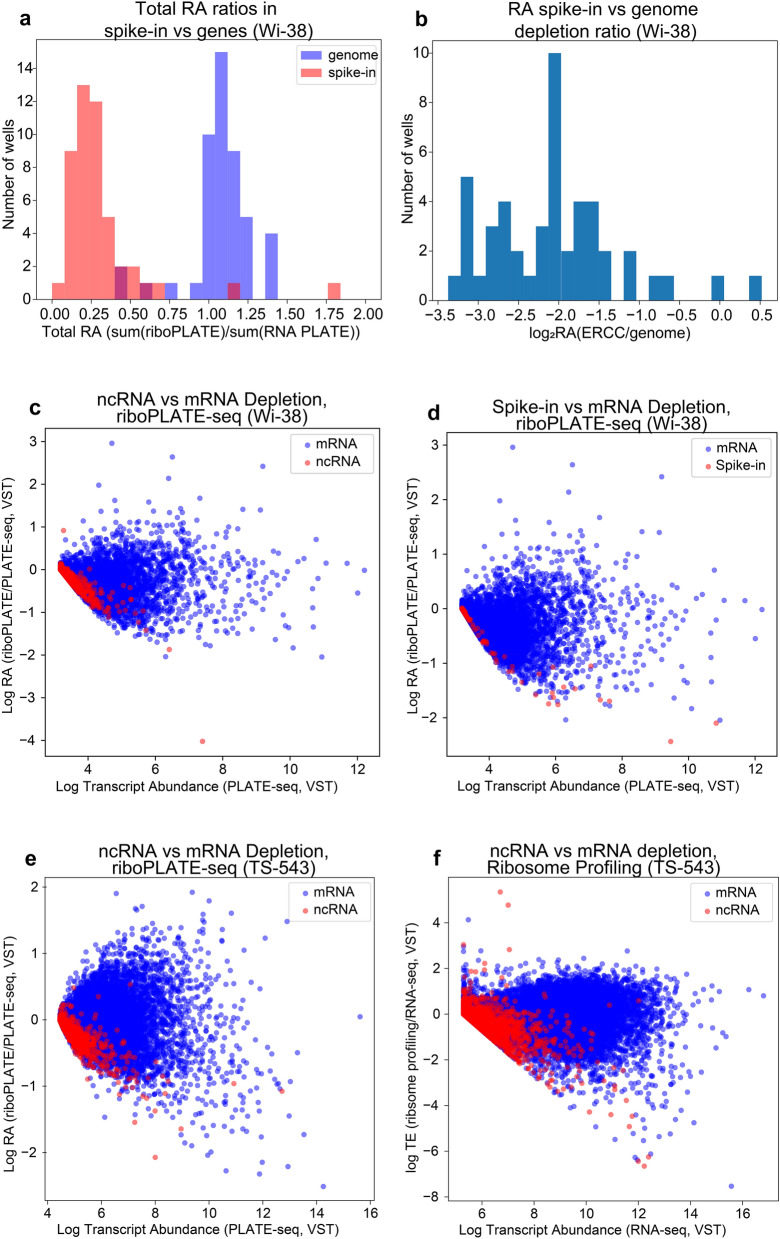
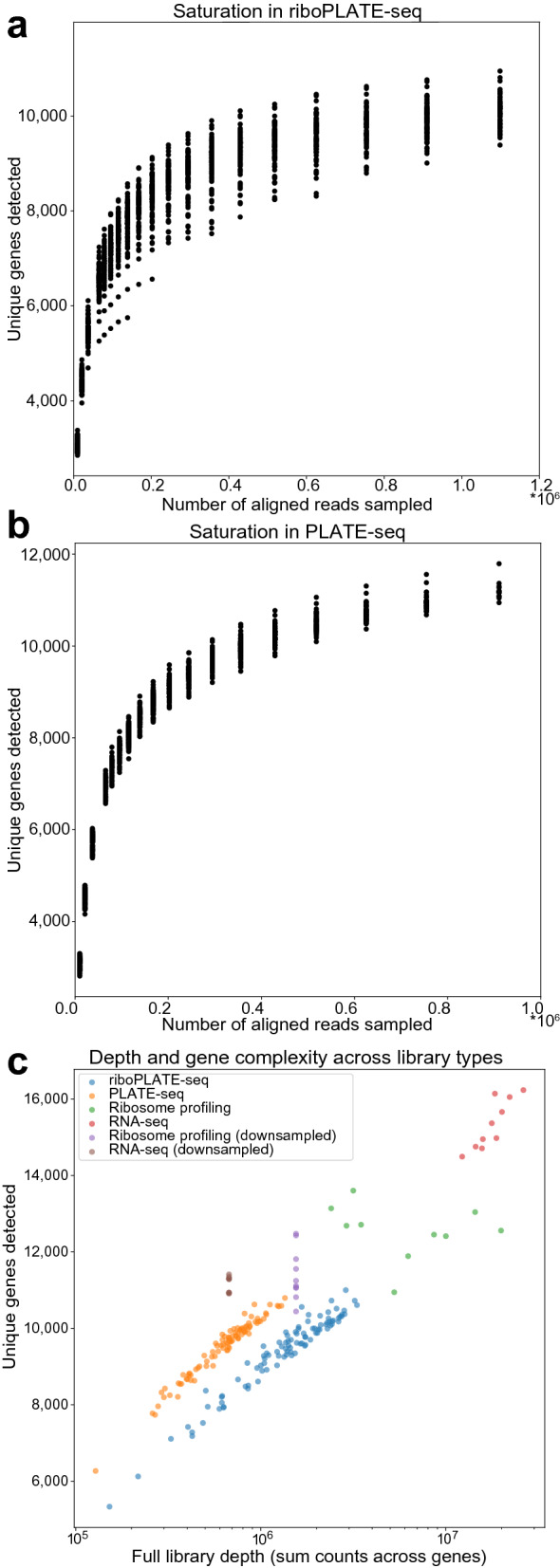
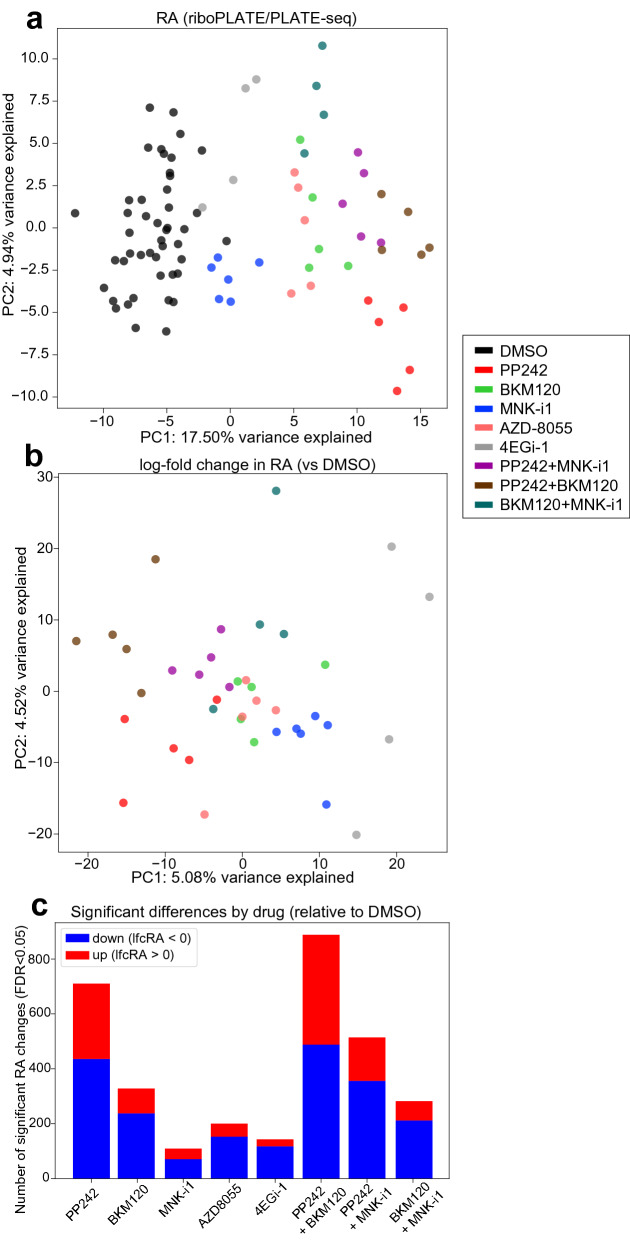
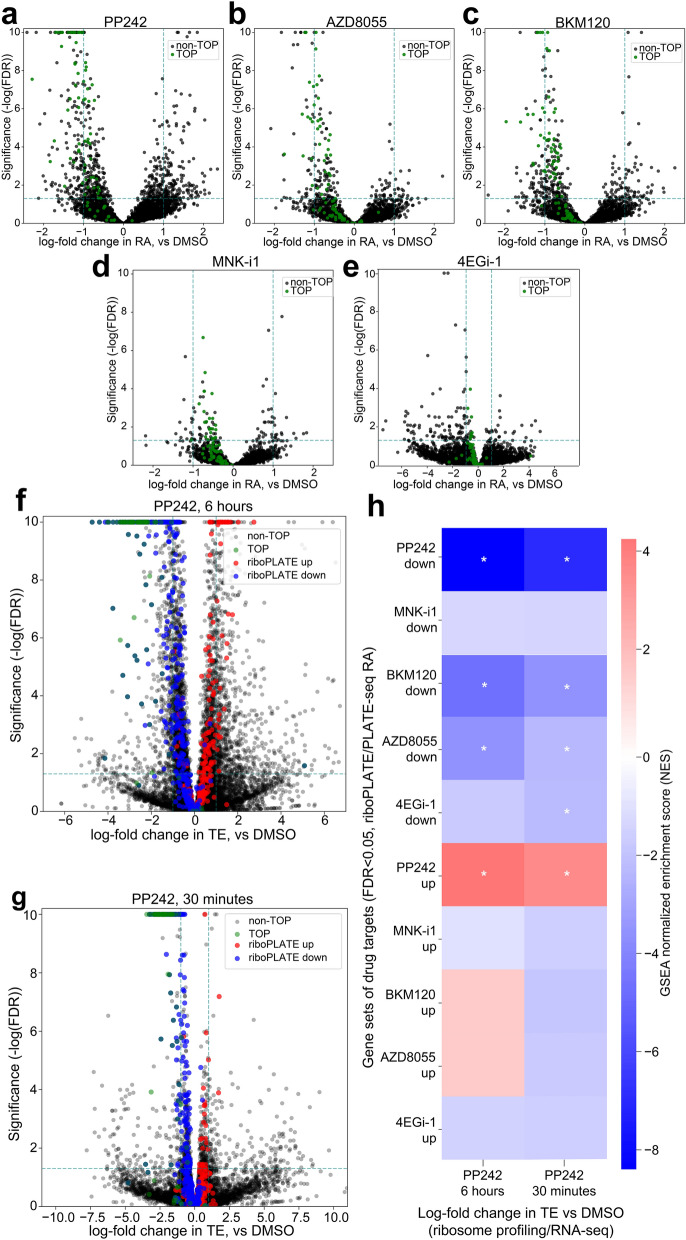
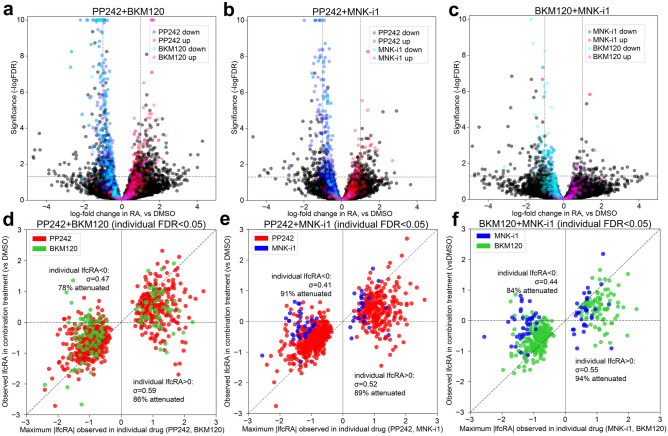
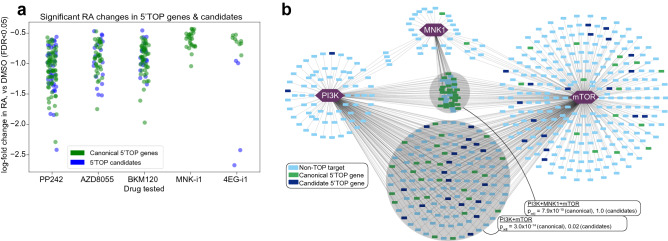
Protein synthesis is dysregulated in many diseases, but we lack a systems-level picture of how signaling molecules and RNA binding proteins interact with the translational machinery, largely due to technological limitations. Here we present riboPLATE-seq, a scalable method for generating paired libraries of ribosome-associated and total mRNA. As an extension of the PLATE-seq protocol, riboPLATE-seq utilizes barcoded primers for pooled library preparation, but additionally leverages anti-rRNA ribosome immunoprecipitation on whole polysomes to measure ribosome association (RA). We compare RA to its analogue in ribosome profiling and RNA sequencing, translation efficiency, and demonstrate both the performance of riboPLATE-seq and its utility in detecting translational alterations induced by specific inhibitors of protein kinases.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
