

Muscle LIM Protein Force-Sensing Mediates Sarcomeric Biomechanical Signaling in Human Familial Hypertrophic Cardiomyopathy
- PMID: 35384713
- PMCID: PMC9109819
- DOI: 10.1161/CIRCULATIONAHA.121.056265
Muscle LIM Protein Force-Sensing Mediates Sarcomeric Biomechanical Signaling in Human Familial Hypertrophic Cardiomyopathy
Abstract
Background: Familial hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac disease and is typically caused by mutations in genes encoding sarcomeric proteins that regulate cardiac contractility. HCM manifestations include left ventricular hypertrophy and heart failure, arrythmias, and sudden cardiac death. How dysregulated sarcomeric force production is sensed and leads to pathological remodeling remains poorly understood in HCM, thereby inhibiting the efficient development of new therapeutics.
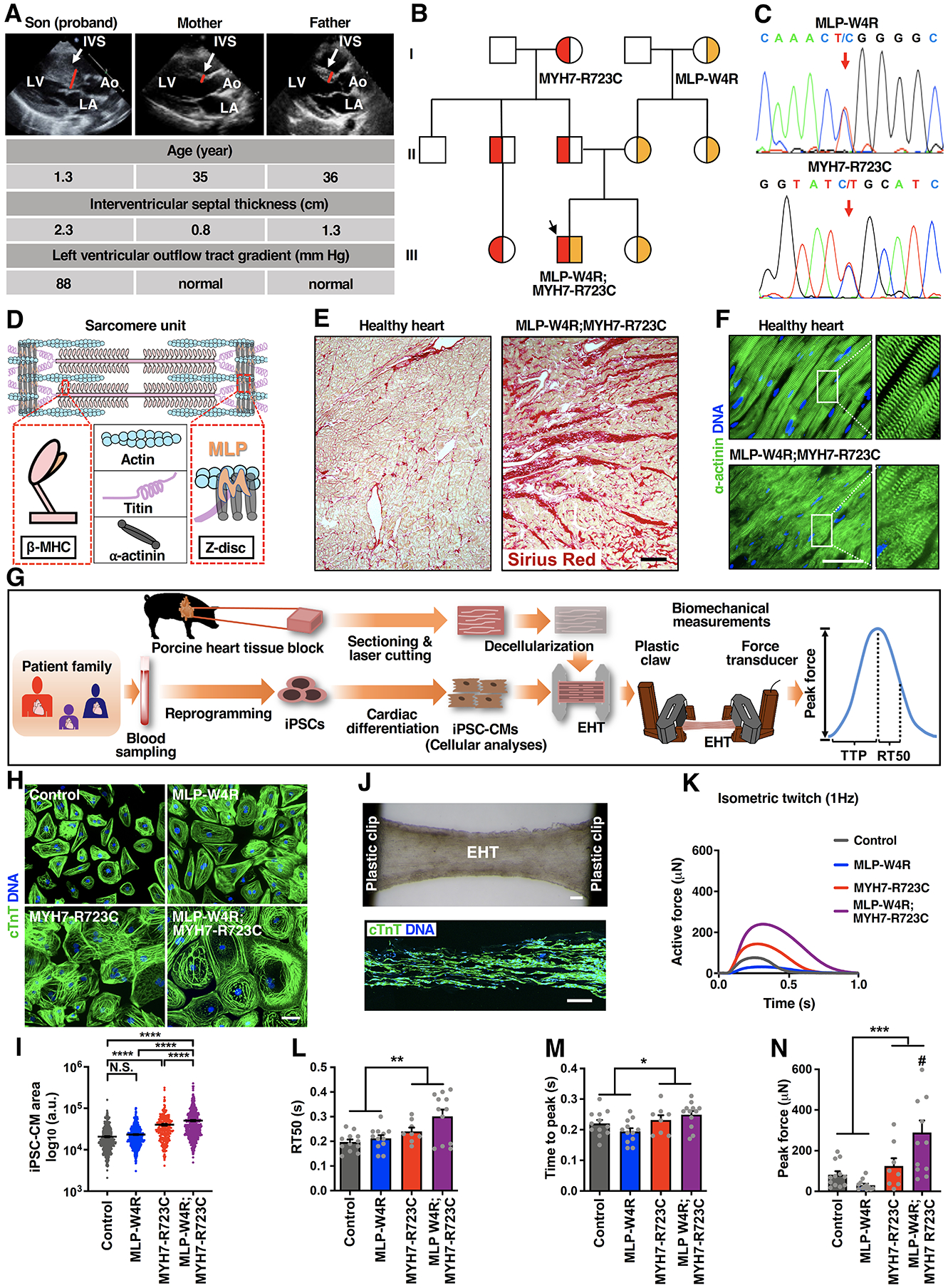
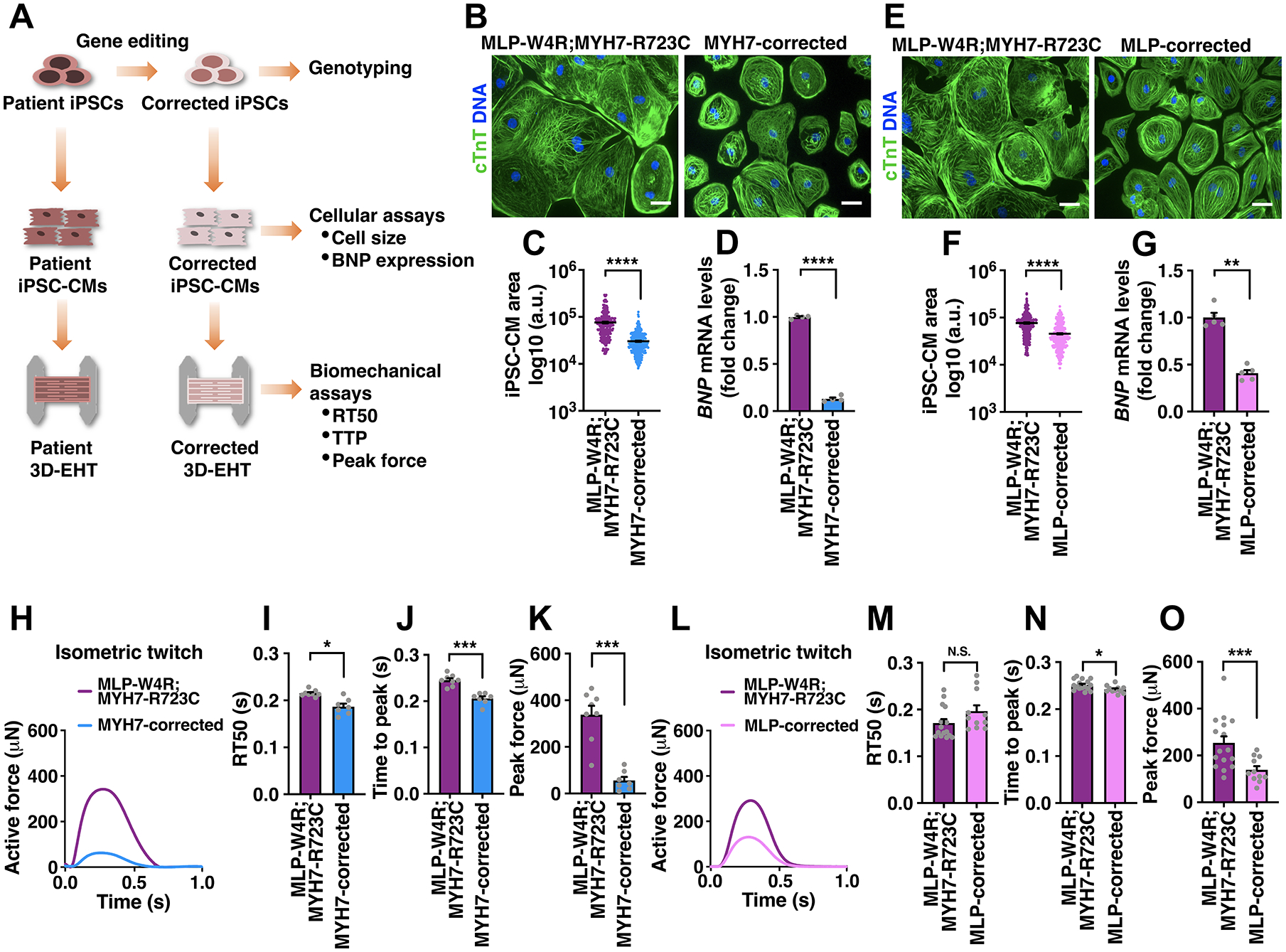
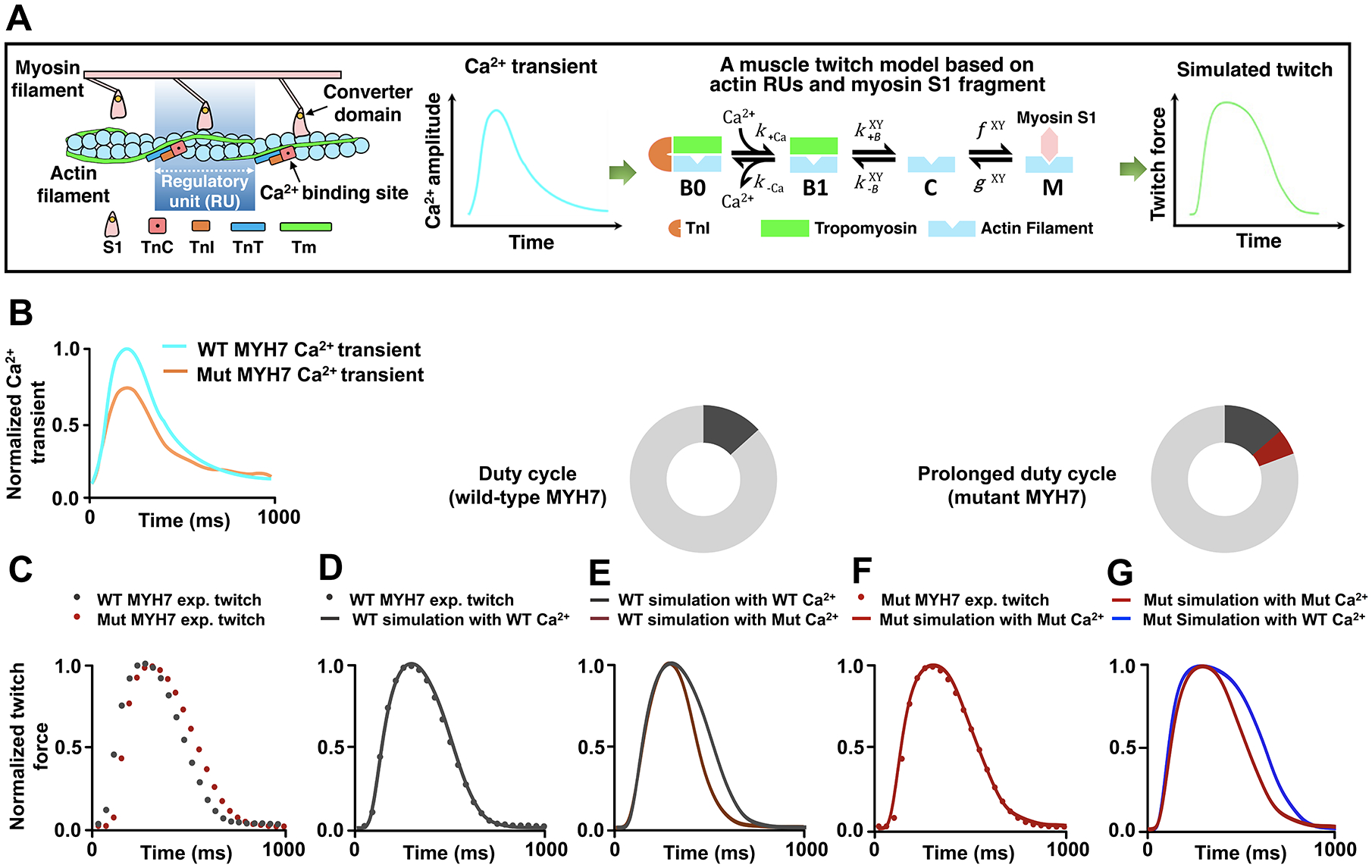
Methods: Our discovery was based on insights from a severe phenotype of an individual with HCM and a second genetic alteration in a sarcomeric mechanosensing protein. We derived cardiomyocytes from patient-specific induced pluripotent stem cells and developed robust engineered heart tissues by seeding induced pluripotent stem cell-derived cardiomyocytes into a laser-cut scaffold possessing native cardiac fiber alignment to study human cardiac mechanobiology at both the cellular and tissue levels. Coupled with computational modeling for muscle contraction and rescue of disease phenotype by gene editing and pharmacological interventions, we have identified a new mechanotransduction pathway in HCM, shown to be essential in modulating the phenotypic expression of HCM in 5 families bearing distinct sarcomeric mutations.
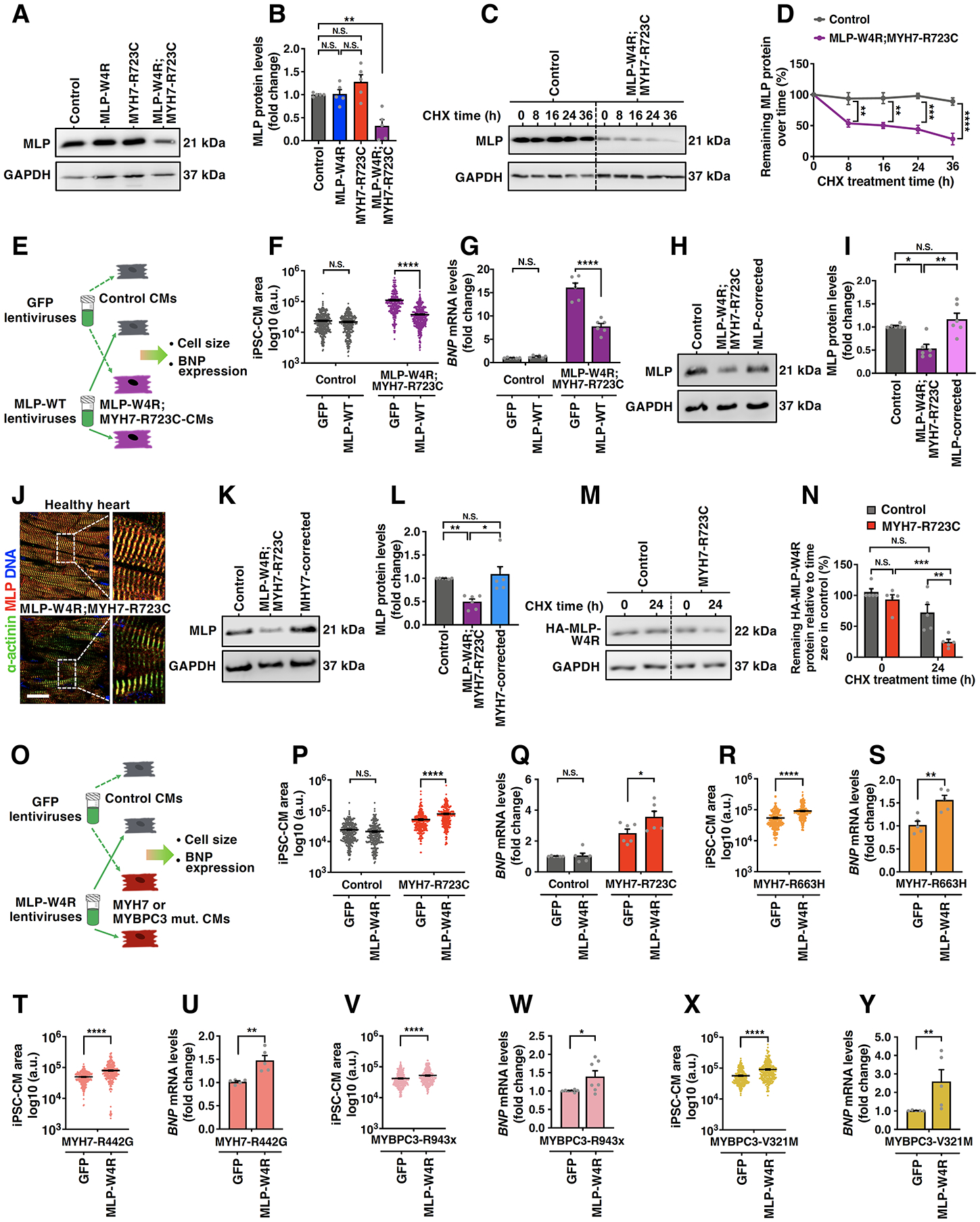
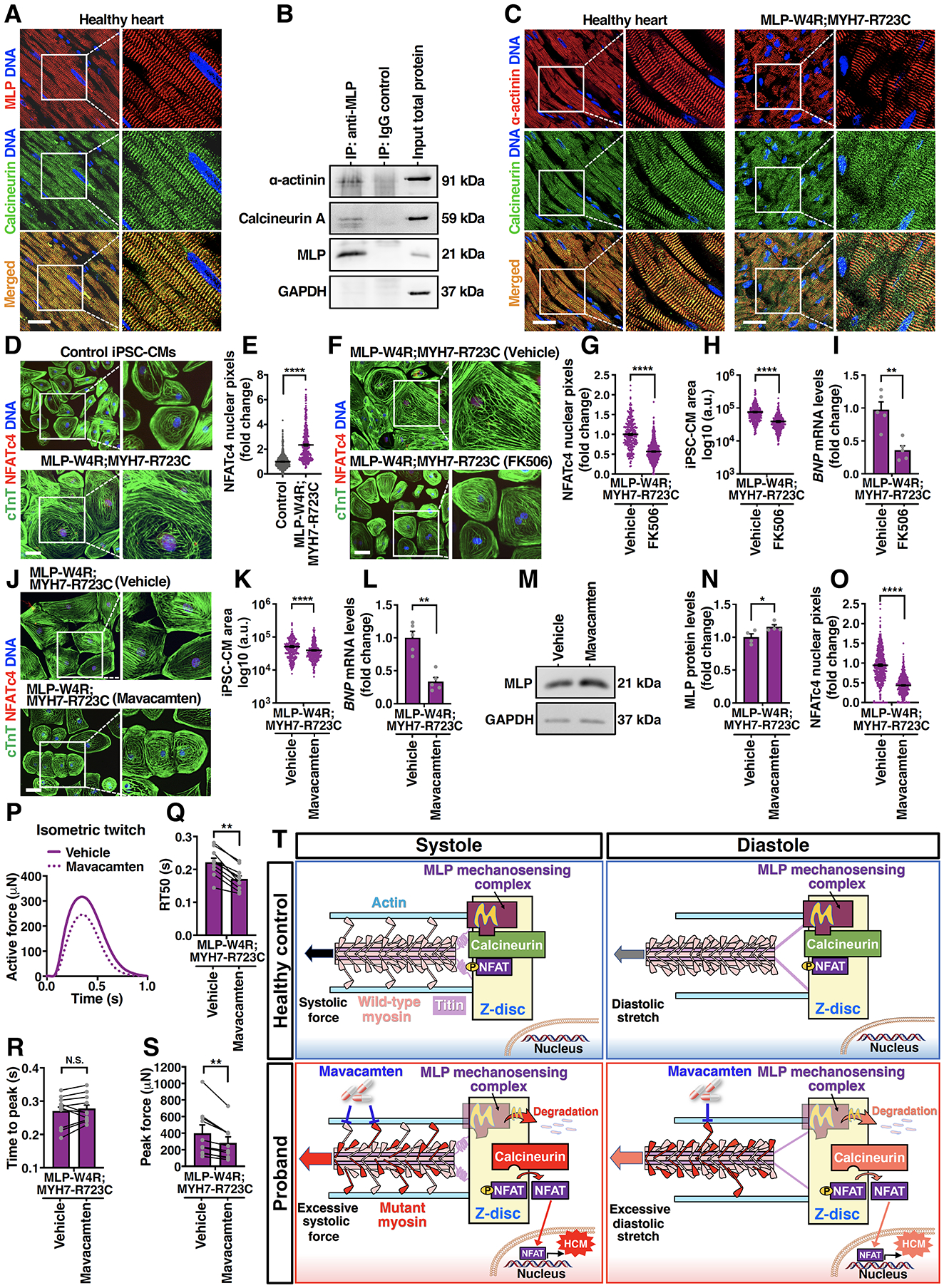
Results: Enhanced actomyosin crossbridge formation caused by sarcomeric mutations in cardiac myosin heavy chain (MYH7) led to increased force generation, which, when coupled with slower twitch relaxation, destabilized the MLP (muscle LIM protein) stretch-sensing complex at the Z-disc. Subsequent reduction in the sarcomeric muscle LIM protein level caused disinhibition of calcineurin-nuclear factor of activated T-cells signaling, which promoted cardiac hypertrophy. We demonstrate that the common muscle LIM protein-W4R variant is an important modifier, exacerbating the phenotypic expression of HCM, but alone may not be a disease-causing mutation. By mitigating enhanced actomyosin crossbridge formation through either genetic or pharmacological means, we alleviated stress at the Z-disc, preventing the development of hypertrophy associated with sarcomeric mutations.
Conclusions: Our studies have uncovered a novel biomechanical mechanism through which dysregulated sarcomeric force production is sensed and leads to pathological signaling, remodeling, and hypertrophic responses. Together, these establish the foundation for developing innovative mechanism-based treatments for HCM that stabilize the Z-disc MLP-mechanosensory complex.
Keywords: cardiomyocytes; cellular mechanotransduction; computer simulation; heart failure; hypertrophic cardiomyopathy; induced pluripotent stem cells; modifier genes.
Conflict of interest statement
Conflict of Interest Disclosures
S.G.C. has equity ownership in Propria LLC, which has licensed technology employed in the studies reported in this manuscript. Propria LLC did not fund these studies, and Propria LLC did not influence the conduct, description, or interpretation of the findings in this report. None of the other authors declares any competing financial interest.
Figures
References
-
- Sen-Chowdhry S, Jacoby D, Moon JC and McKenna WJ. Update on hypertrophic cardiomyopathy and a guide to the guidelines. Nat Rev Cardiol. 2016;13:651–675. - PubMed
-
- Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, Caleshu CA, Helms AS, Colan SD, Girolami F, Cecchi F, Seidman CE, Sajeev G, Signorovitch J, Green EM and Olivotto I. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018;138:1387–1398. - PMC - PubMed
-
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–20. - PubMed
-
- Ashrafian H and Watkins H. Reviews of translational medicine and genomics in cardiovascular disease: new disease taxonomy and therapeutic implications cardiomyopathies: therapeutics based on molecular phenotype. J Am Coll Cardiol. 2007;49:1251–64. - PubMed
-
- Tardiff JC. Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev. 2005;10:237–48. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
