

Metabolism drives macrophage heterogeneity in the tumor microenvironment
- PMID: 35385733
- PMCID: PMC9052943
- DOI: 10.1016/j.celrep.2022.110609
Metabolism drives macrophage heterogeneity in the tumor microenvironment
Abstract
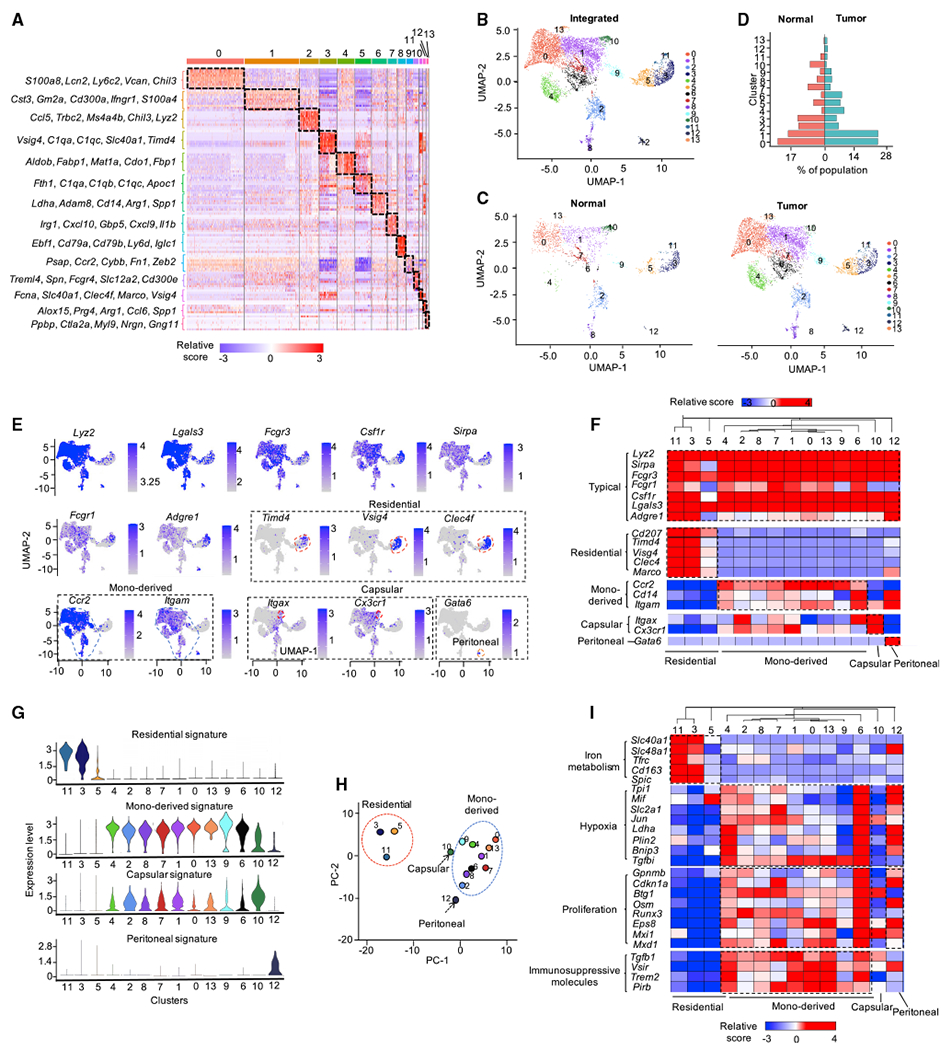
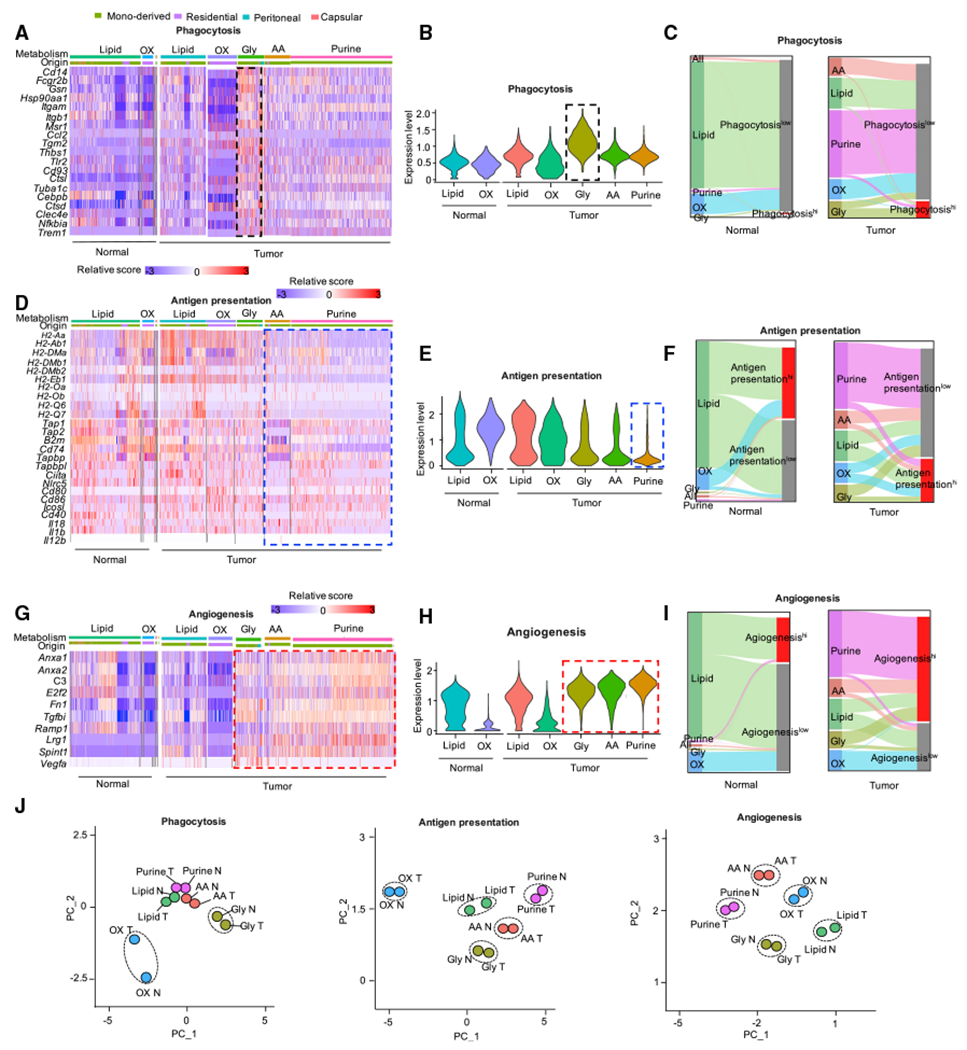
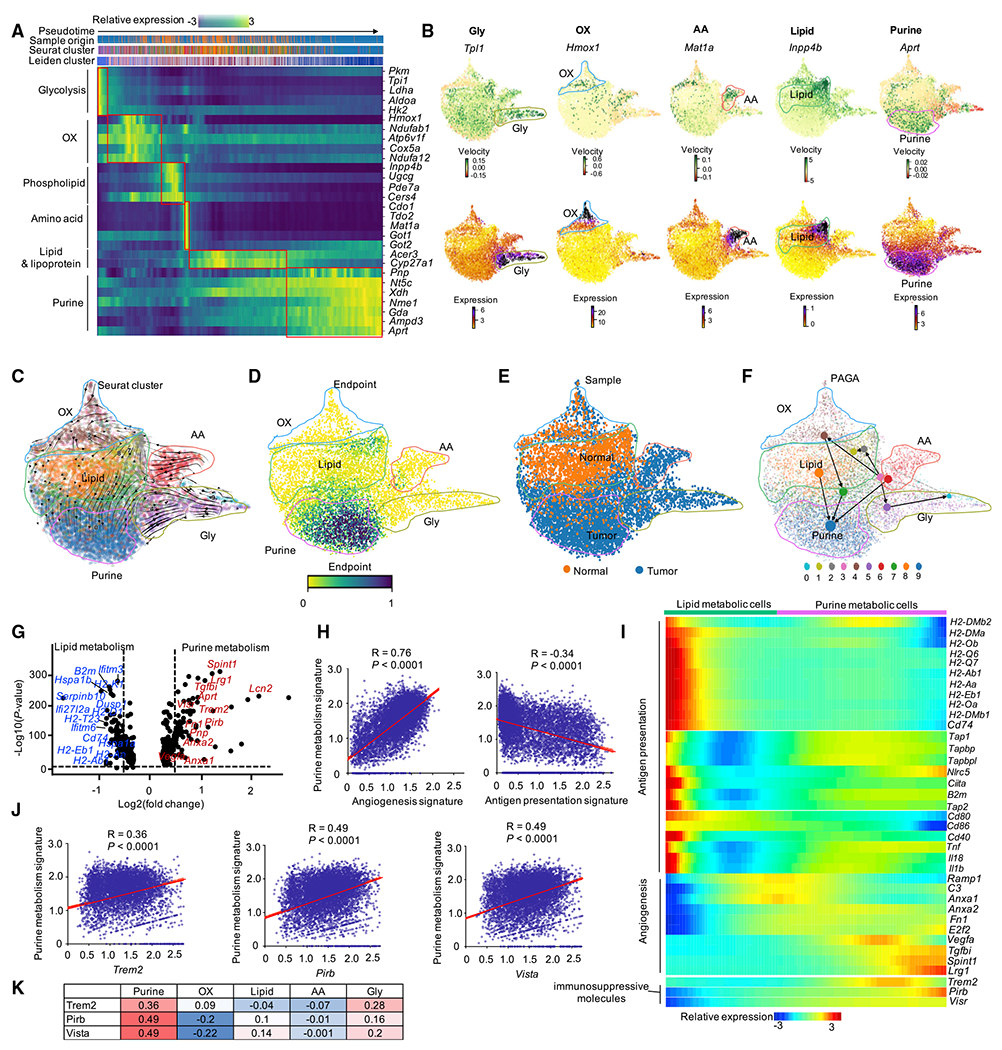
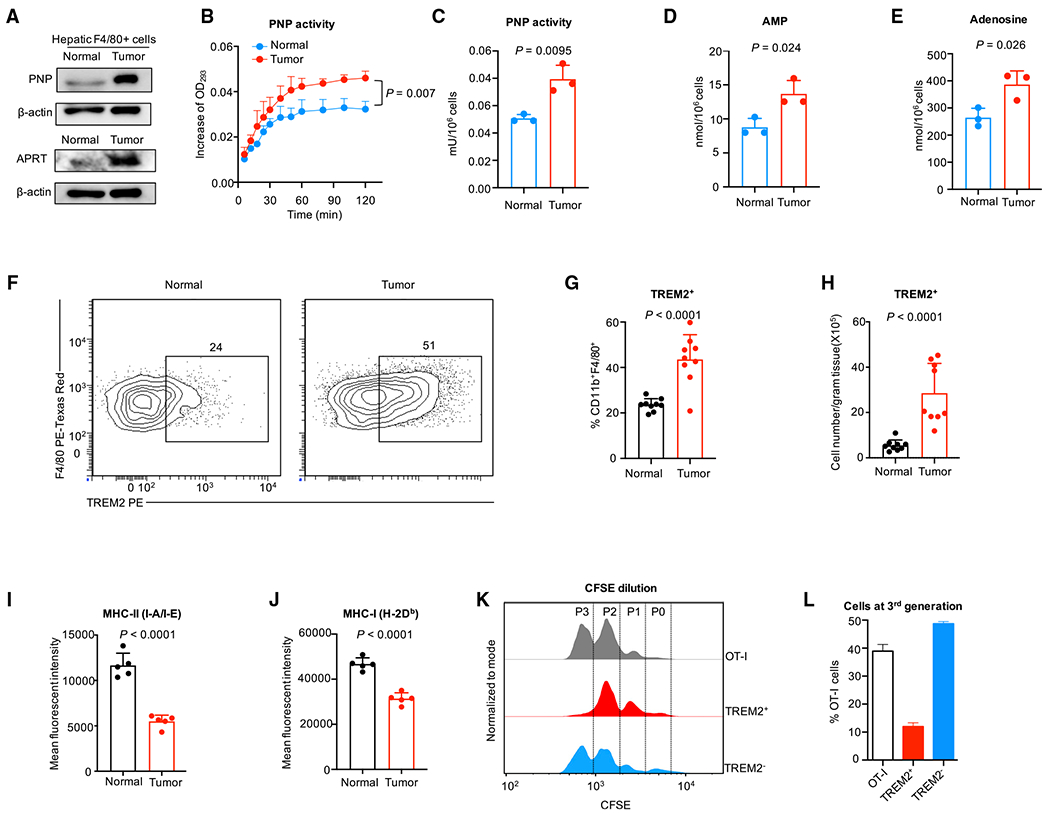
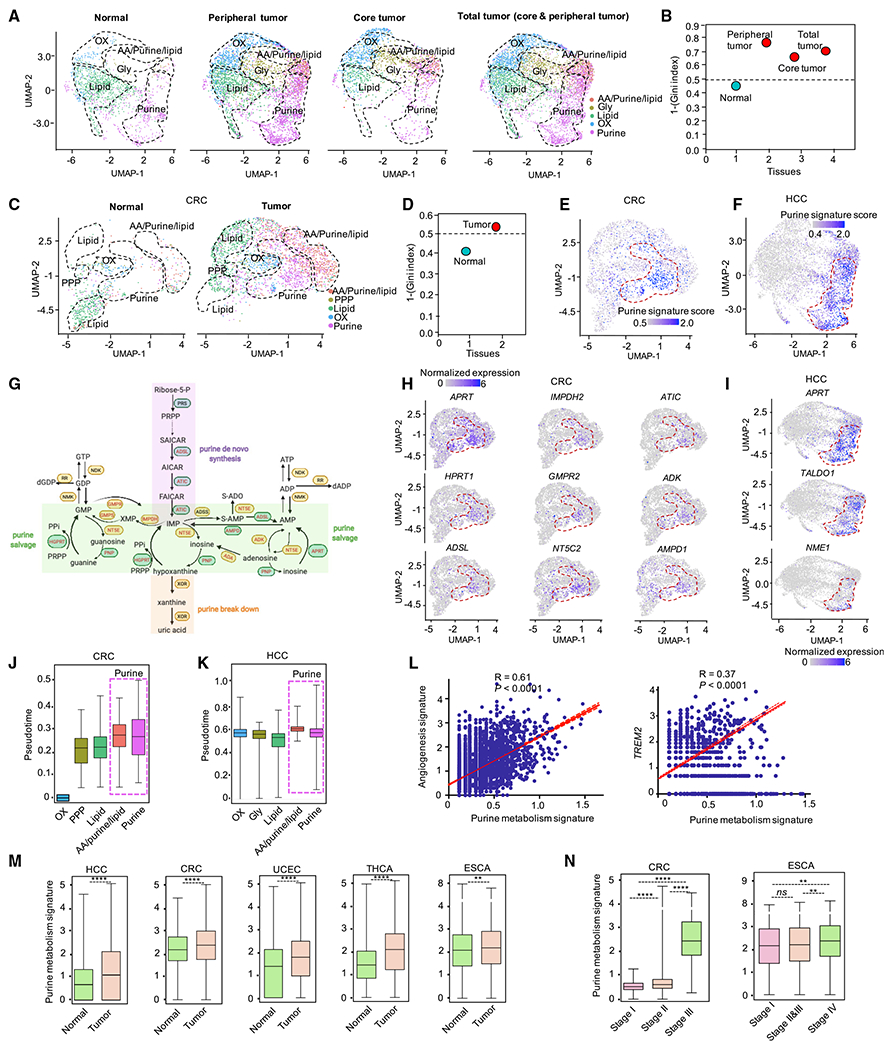
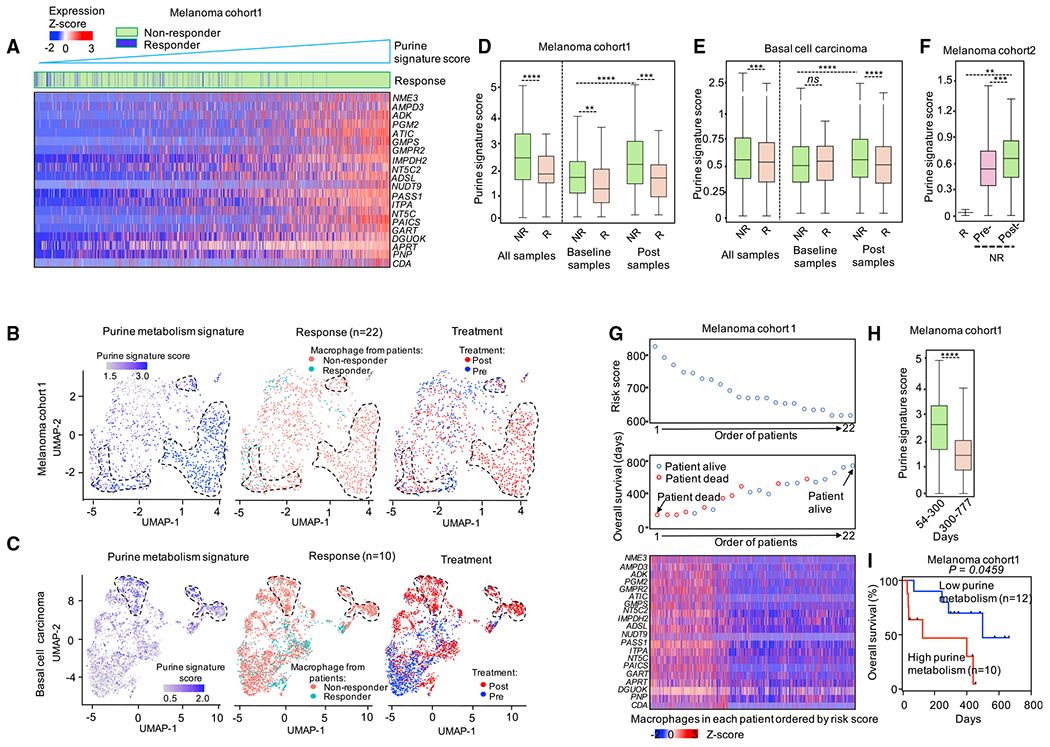
Tumor-associated macrophages (TAMs) are a major cellular component in the tumor microenvironment (TME). However, the relationship between the phenotype and metabolic pattern of TAMs remains poorly understood. We performed single-cell transcriptome profiling on hepatic TAMs from mice bearing liver metastatic tumors. We find that TAMs manifest high heterogeneity at the levels of transcription, development, metabolism, and function. Integrative analyses and validation experiments indicate that increased purine metabolism is a feature of TAMs with pro-tumor and terminal differentiation phenotypes. Like mouse TAMs, human TAMs are highly heterogeneous. Human TAMs with increased purine metabolism exhibit a pro-tumor phenotype and correlate with poor therapeutic efficacy to immune checkpoint blockade. Altogether, our work demonstrates that TAMs are developmentally, metabolically, and functionally heterogeneous and purine metabolism may be a key metabolic feature of a pro-tumor macrophage population.
Keywords: CP: Cancer; CP: Metabolism; cancer; checkpoint; immunosuppression; immunotherapy; liver; macrophage; metabolism; purine; single-cell RNA sequencing; tumor microenvironment.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests We declare that we have no competing interests.
Figures

References
-
- Ben-Moshe S, and Itzkovitz S (2019). Spatial heterogeneity in the mammalian liver. Nat. Rev. Gastroenterol. Hepatol 16, 395–410. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
