

Redox signaling at the crossroads of human health and disease
- PMID: 35386842
- PMCID: PMC8971743
- DOI: 10.1002/mco2.127
Redox signaling at the crossroads of human health and disease
Abstract
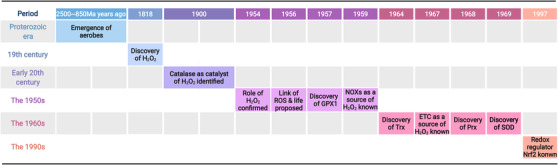
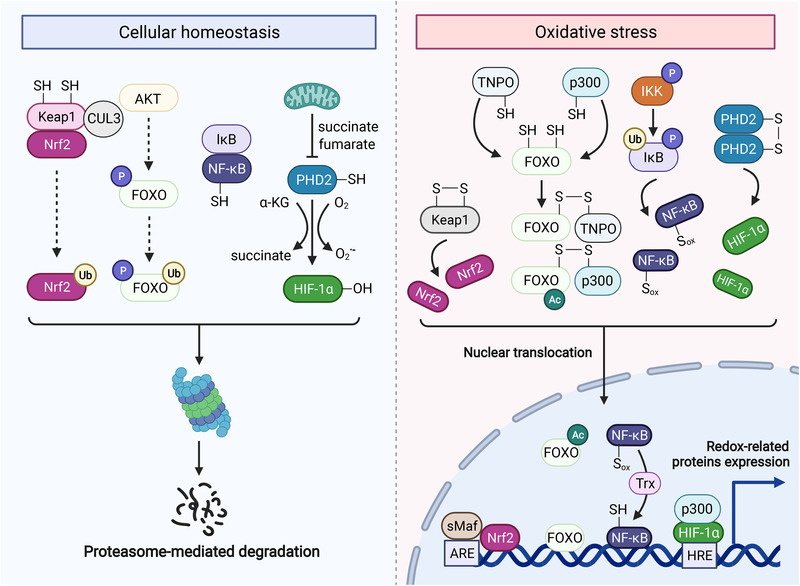
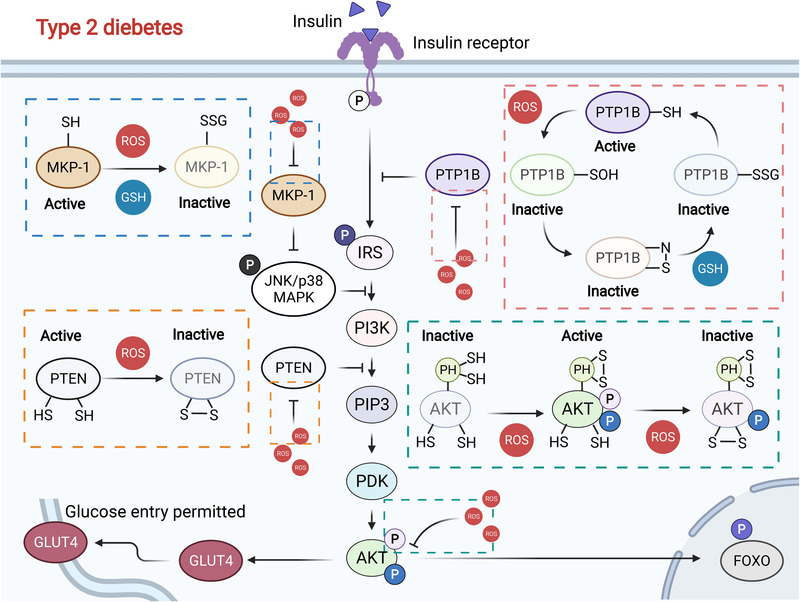
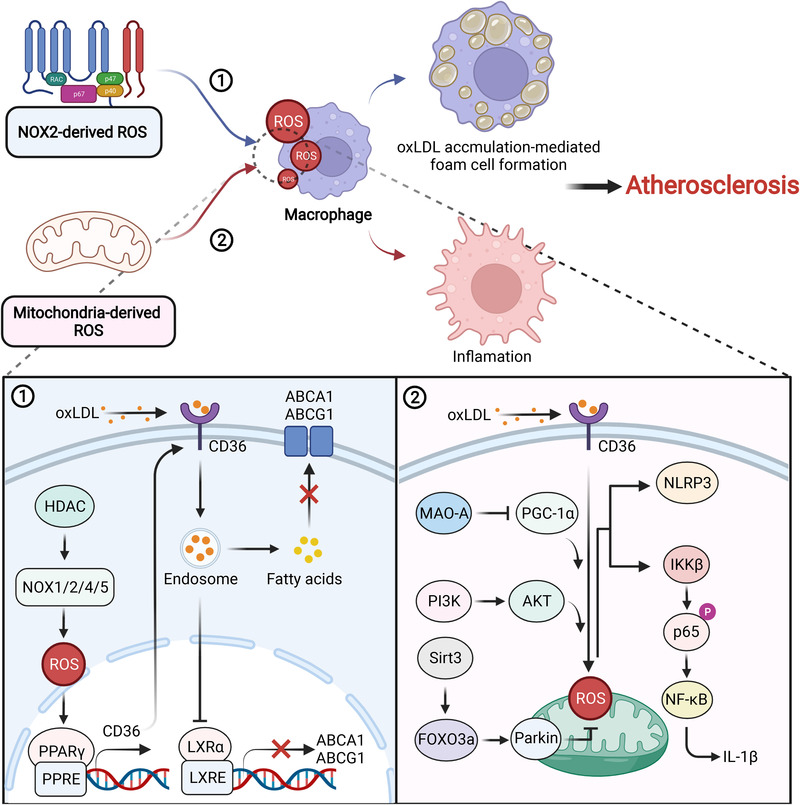
Redox biology is at the core of life sciences, accompanied by the close correlation of redox processes with biological activities. Redox homeostasis is a prerequisite for human health, in which the physiological levels of nonradical reactive oxygen species (ROS) function as the primary second messengers to modulate physiological redox signaling by orchestrating multiple redox sensors. However, excessive ROS accumulation, termed oxidative stress (OS), leads to biomolecule damage and subsequent occurrence of various diseases such as type 2 diabetes, atherosclerosis, and cancer. Herein, starting with the evolution of redox biology, we reveal the roles of ROS as multifaceted physiological modulators to mediate redox signaling and sustain redox homeostasis. In addition, we also emphasize the detailed OS mechanisms involved in the initiation and development of several important diseases. ROS as a double-edged sword in disease progression suggest two different therapeutic strategies to treat redox-relevant diseases, in which targeting ROS sources and redox-related effectors to manipulate redox homeostasis will largely promote precision medicine. Therefore, a comprehensive understanding of the redox signaling networks under physiological and pathological conditions will facilitate the development of redox medicine and benefit patients with redox-relevant diseases.
Keywords: hydrogen peroxide; oxidative stress; reactive oxygen species; redox signaling; redox therapy; redox‐relevant disease.
© 2022 The Authors. MedComm published by Sichuan International Medical Exchange & Promotion Association (SCIMEA) and John Wiley & Sons Australia, Ltd.
Conflict of interest statement
Canhua Huang is an editorial board member of MedComm. Author Canhua Huang was not involved in the journal's review of, or decisions related to, this manuscript. The other authors have no conflicts of interest to declare.
Figures
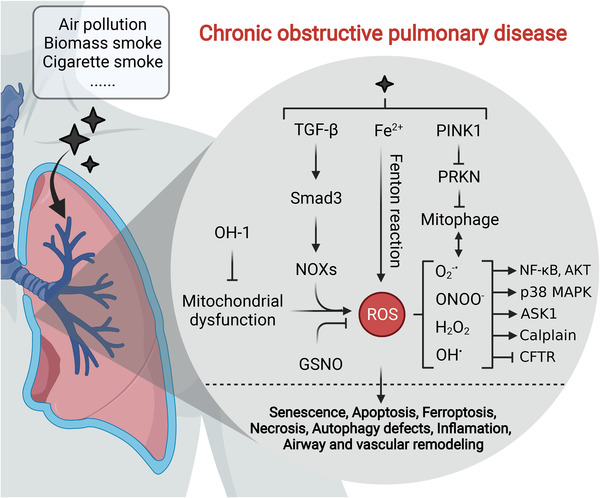
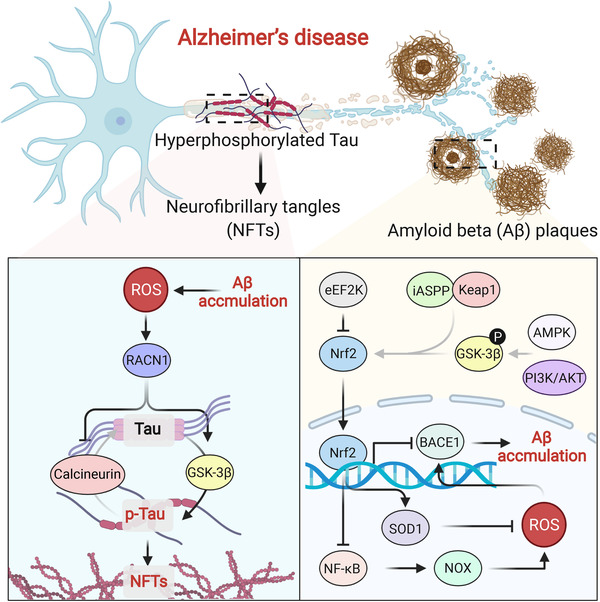
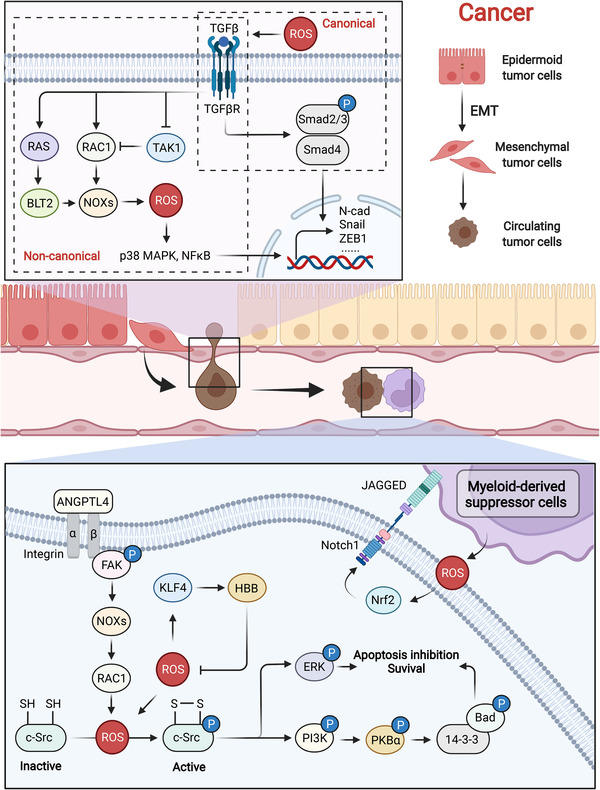
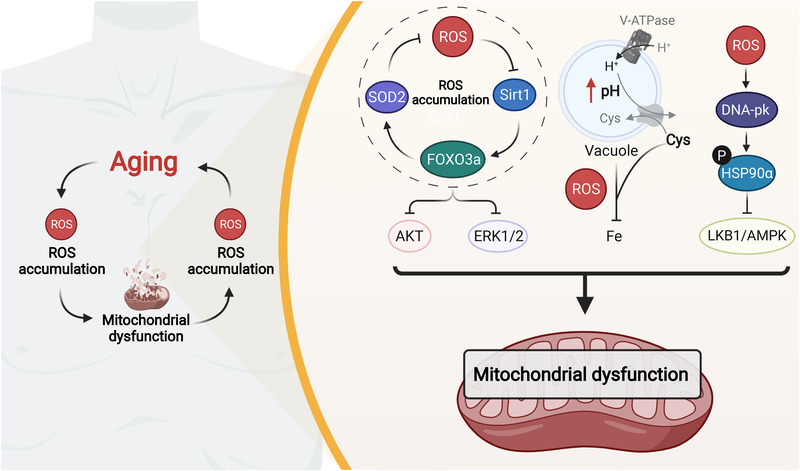
References
-
- Herrmann JM, Dick TP. Redox biology on the rise. Biol Chem. 2012;393(9):999‐1004. - PubMed
-
- Mantzaris MD, Bellou S, Skiada V, Kitsati N, Fotsis T, Galaris D. Intracellular labile iron determines H2O2‐induced apoptotic signaling via sustained activation of ASK1/JNK‐p38 axis. Free Radic Biol Med. 2016;97:454‐465. - PubMed
Publication types
LinkOut - more resources
Full Text Sources