

scSPLAT, a scalable plate-based protocol for single cell WGBS library preparation
- PMID: 35388090
- PMCID: PMC8986790
- DOI: 10.1038/s41598-022-09798-2
scSPLAT, a scalable plate-based protocol for single cell WGBS library preparation
Abstract
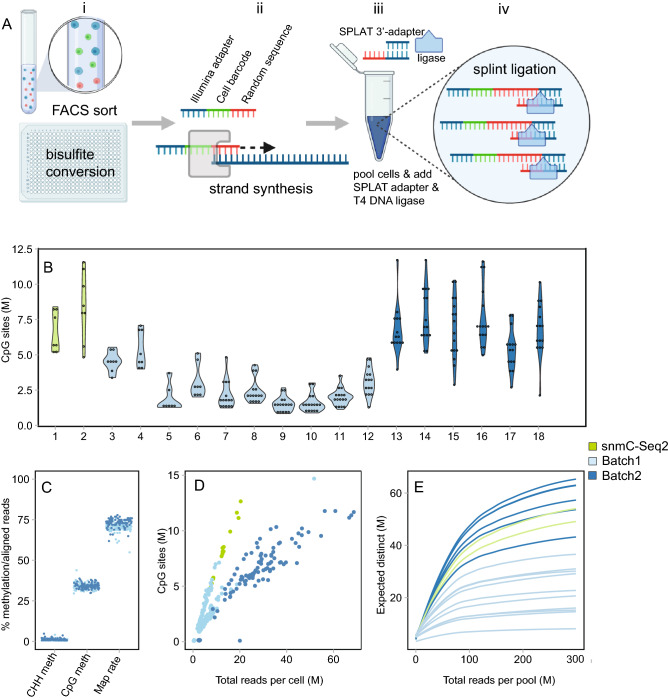
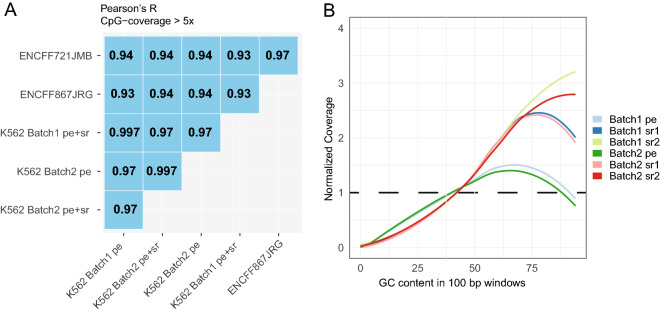
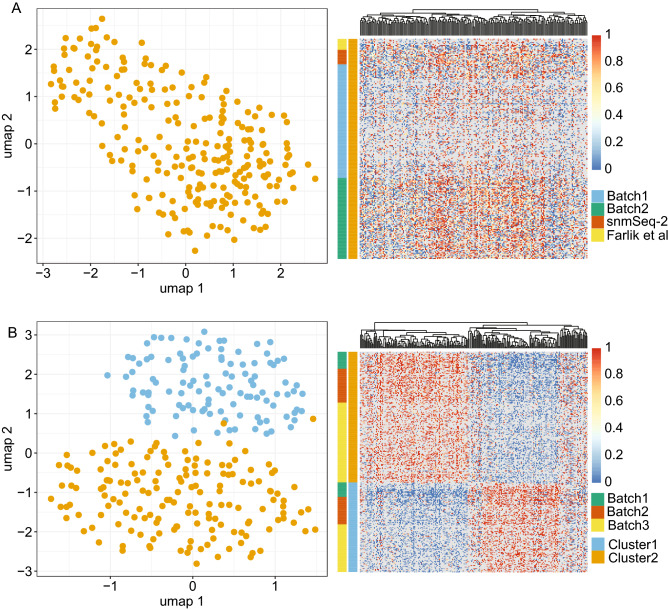
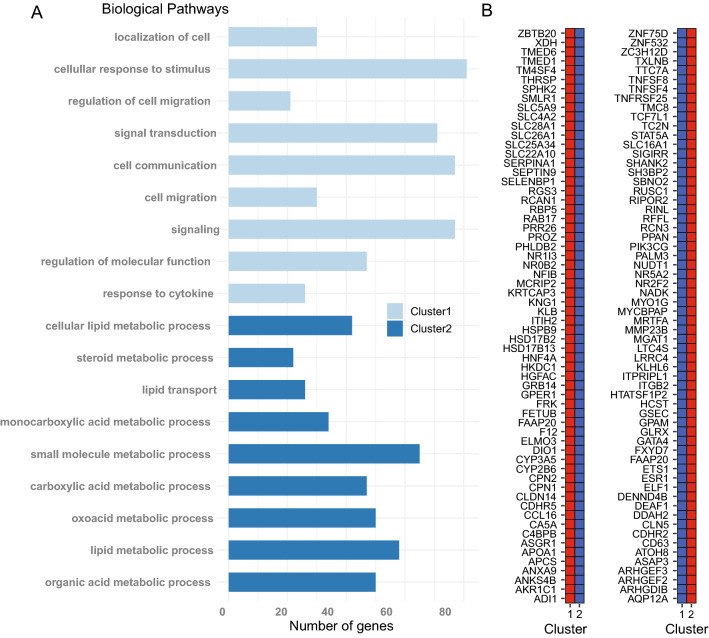
DNA methylation is a central epigenetic mark that has diverse roles in gene regulation, development, and maintenance of genome integrity. 5 methyl cytosine (5mC) can be interrogated at base resolution in single cells by using bisulfite sequencing (scWGBS). Several different scWGBS strategies have been described in recent years to study DNA methylation in single cells. However, there remain limitations with respect to cost-efficiency and yield. Herein, we present a new development in the field of scWGBS library preparation; single cell Splinted Ligation Adapter Tagging (scSPLAT). scSPLAT employs a pooling strategy to facilitate sample preparation at a higher scale and throughput than previously possible. We demonstrate the accuracy and robustness of the method by generating data from 225 single K562 cells and from 309 single liver nuclei and compare scSPLAT against other scWGBS methods.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Hui T, Cao Q, Wegrzyn-Woltosz J, O'Neill K, Hammond CA, Knapp DJ, Laks E, Moksa M, Aparicio S, Eaves CJ, Karsan A. High-resolution single-cell DNA methylation measurements reveal epigenetically distinct hematopoietic stem cell subpopulations. Stem Cell Rep. 2018;11:578–592. doi: 10.1016/j.stemcr.2018.07.003. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
