

IL-17-induced HIF1α drives resistance to anti-PD-L1 via fibroblast-mediated immune exclusion
- PMID: 35389431
- PMCID: PMC8996325
- DOI: 10.1084/jem.20210693
IL-17-induced HIF1α drives resistance to anti-PD-L1 via fibroblast-mediated immune exclusion
Abstract
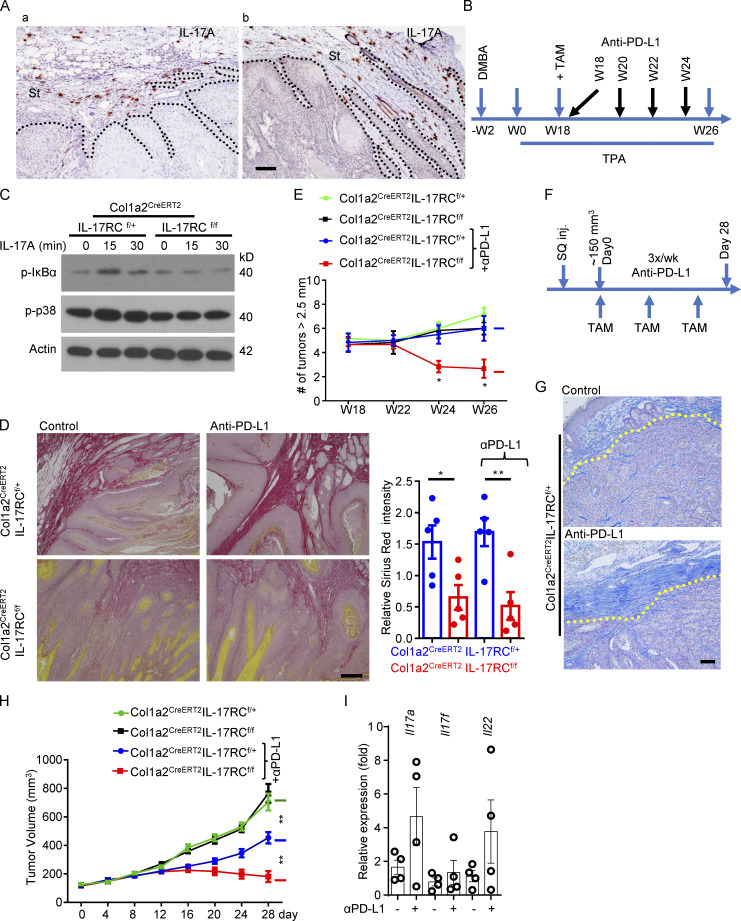
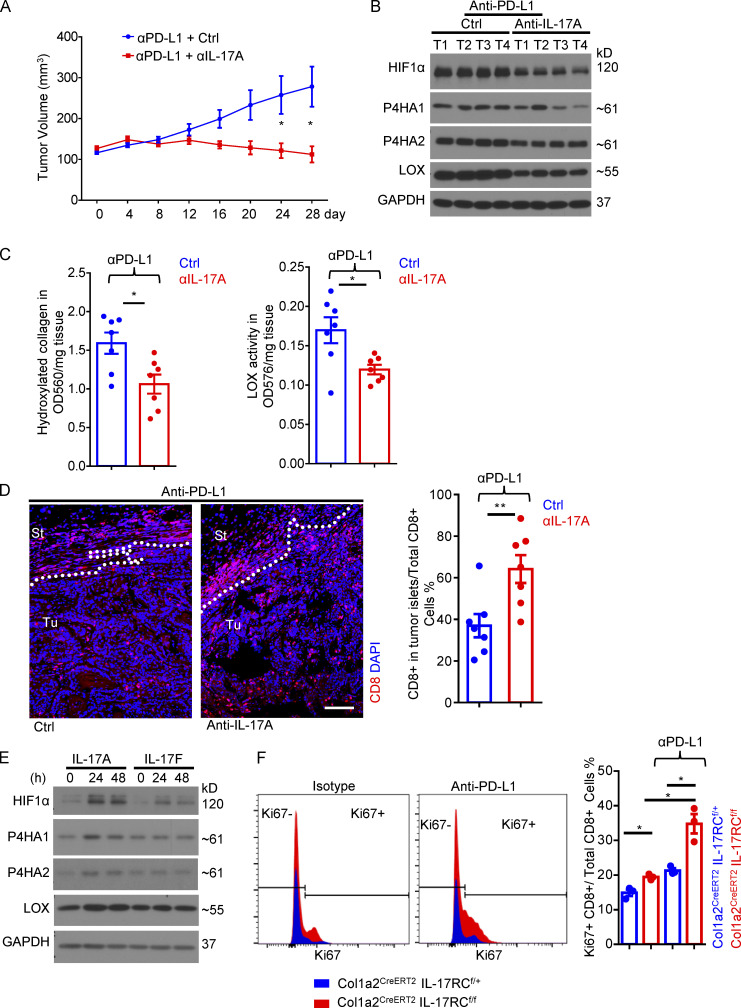
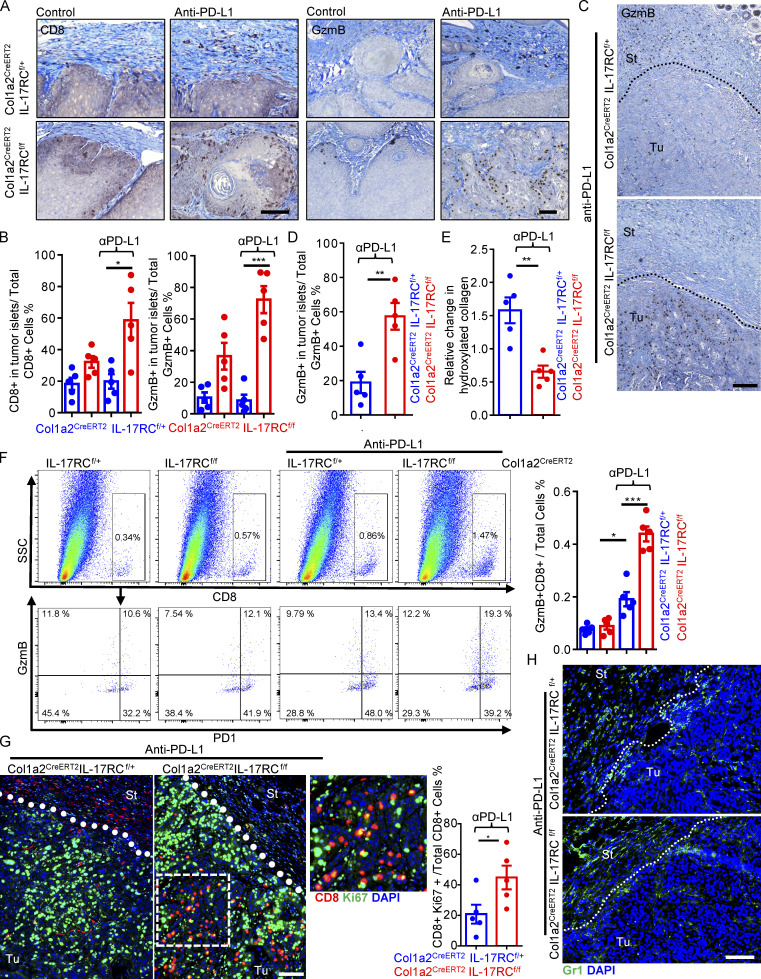
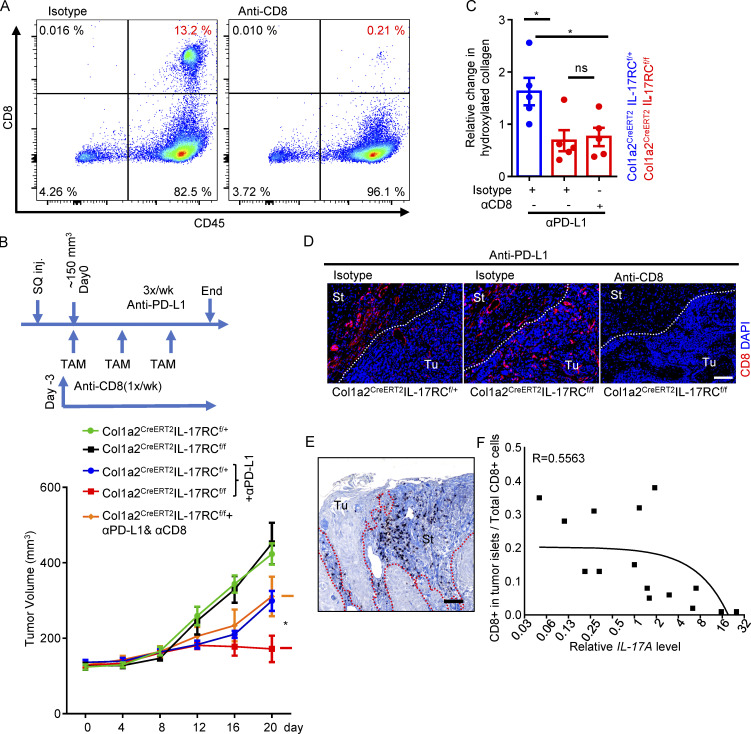
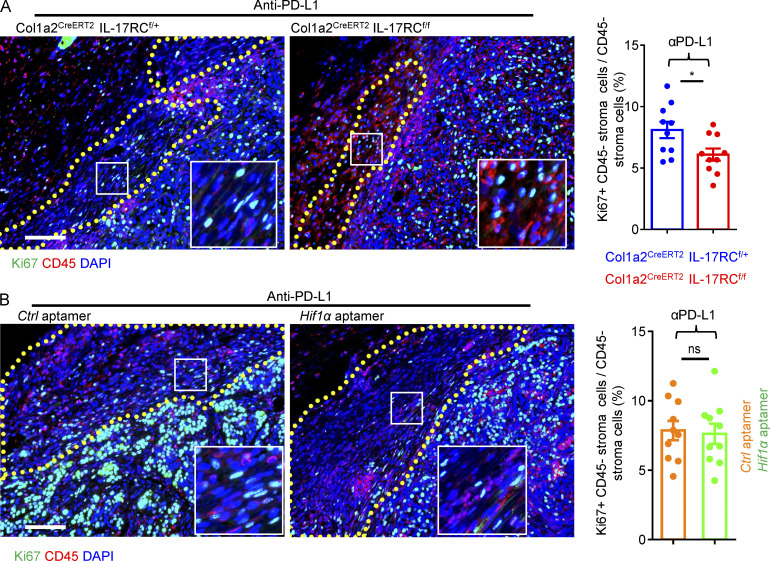
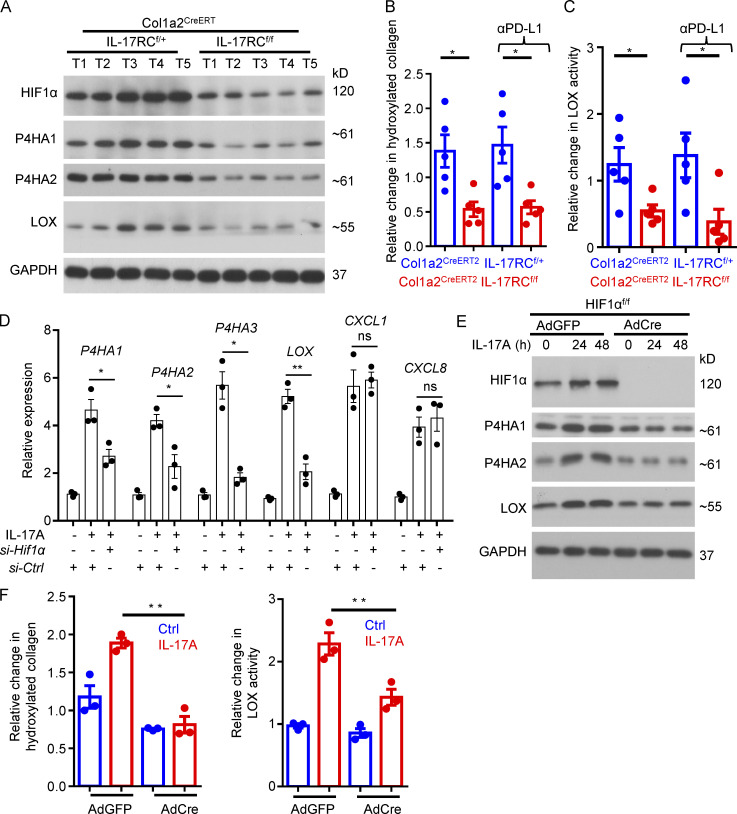
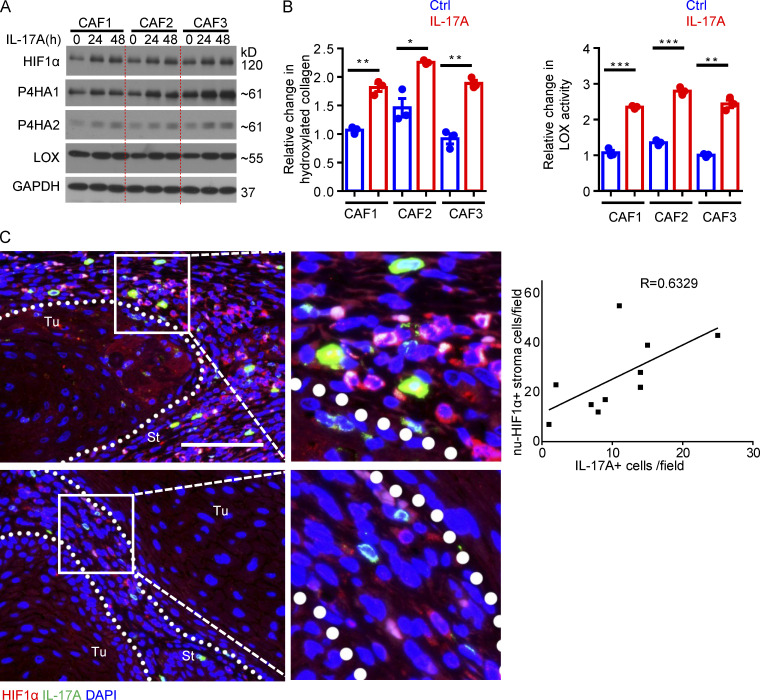
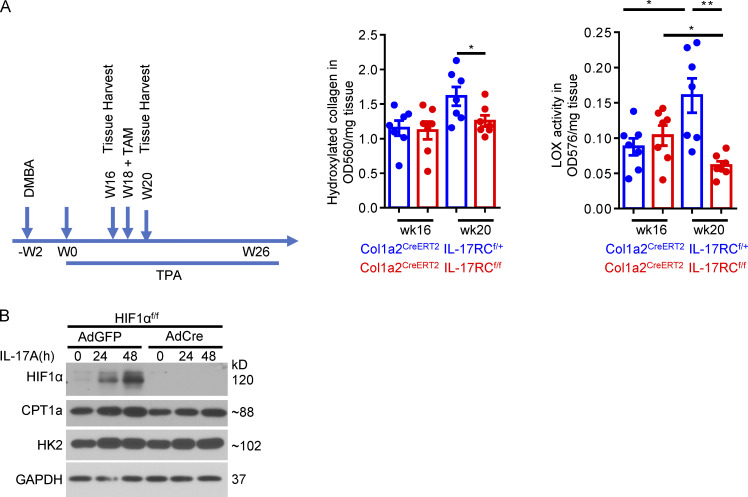
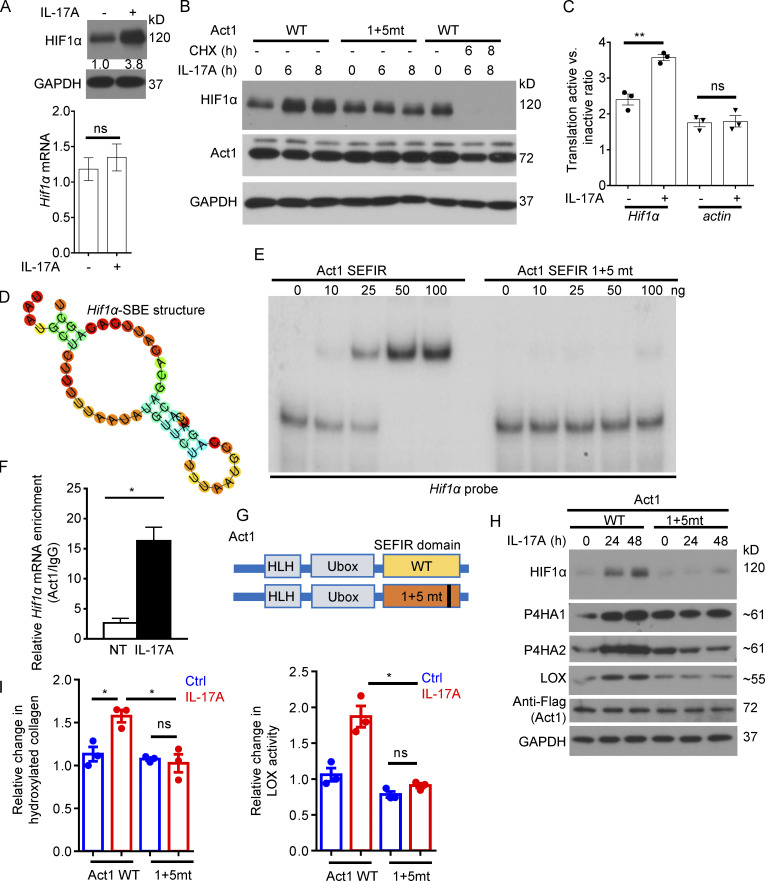
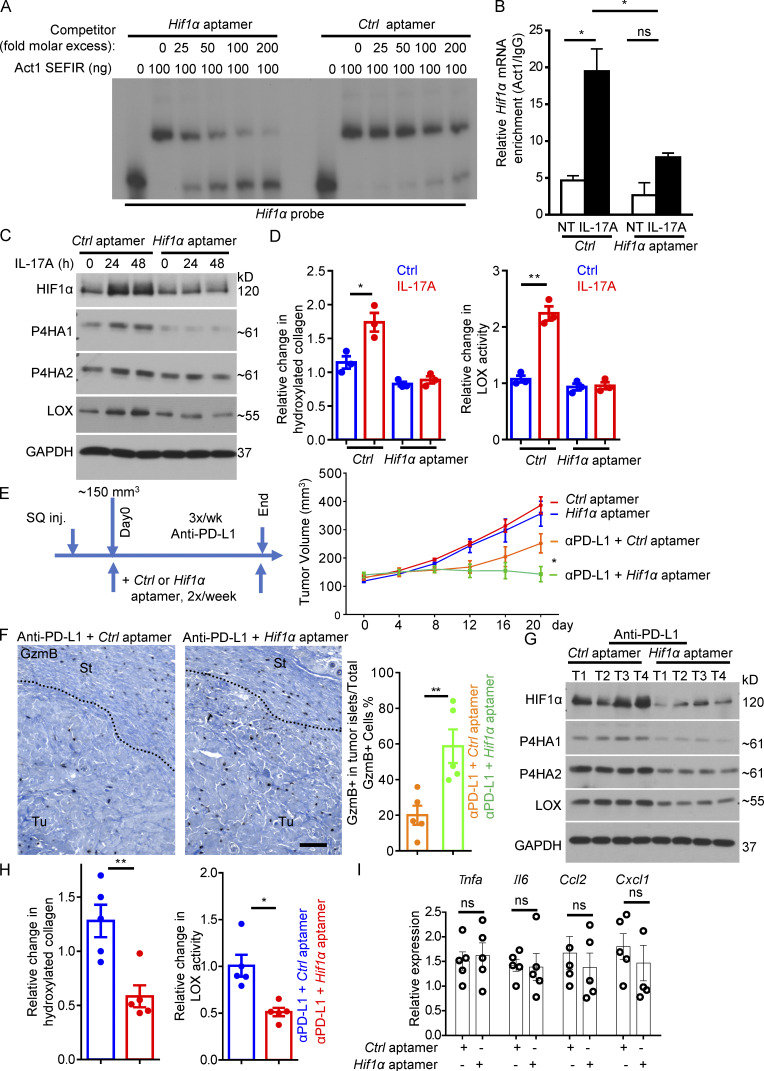
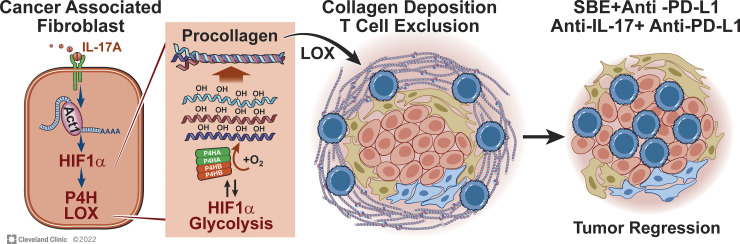
Increasing evidence suggests that intratumoral inflammation has an outsized influence on antitumor immunity. Here, we report that IL-17, a proinflammatory cytokine widely associated with poor prognosis in solid tumors, drives the therapeutic failure of anti-PD-L1. By timing the deletion of IL-17 signaling specifically in cancer-associated fibroblasts (CAFs) in late-stage tumors, we show that IL-17 signaling drives immune exclusion by activating a collagen deposition program in murine models of cutaneous squamous cell carcinoma (cSCC). Ablation of IL-17 signaling in CAFs increased the infiltration of cytotoxic T cells into the tumor mass and sensitized otherwise resistant cSCC to anti-PD-L1 treatment. Mechanistically, the collagen deposition program in CAFs was driven by IL-17-induced translation of HIF1α, which was mediated by direct binding of Act1, the adaptor protein of IL-17 receptor, to a stem-loop structure in the 3' untranslated region (UTR) in Hif1α mRNA. Disruption of Act1's binding to Hif1α mRNA abolished IL-17-induced collagen deposition and enhanced anti-PD-L1-mediated tumor regression.
© 2022 Chen et al.
Conflict of interest statement
Disclosures: T. Herjan, L. Hong, C. Liu, and Xiaoxia Li reported a patent to US20200157544A1 pending, a patent to EP3655007A4 pending, and a patent to CN111093678A pending. No other disclosures were reported.
Figures
Comment in
-
Matrix reboot: IL-17 signals CAFs to create a second tumor T cell checkpoint.J Exp Med. 2022 Jun 6;219(6):e20220444. doi: 10.1084/jem.20220444. Epub 2022 May 18. J Exp Med. 2022. PMID: 35583818 Free PMC article.
References
-
- Akbay, E.A., Koyama S., Liu Y., Dries R., Bufe L.E., Silkes M., Alam M.M., Magee D.M., Jones R., Jinushi M., et al. . 2017. Interleukin-17A promotes lung tumor progression through neutrophil attraction to tumor sites and mediating resistance to PD-1 blockade. J. Thorac. Oncol. 12:1268–1279. 10.1016/j.jtho.2017.04.017 - DOI - PMC - PubMed
-
- Amatya, N., Childs E.E., Cruz J.A., Aggor F.E.Y., Garg A.V., Berman A.J., Gudjonsson J.E., Atasoy U., and Gaffen S.L.. 2018. IL-17 integrates multiple self-reinforcing, feed-forward mechanisms through the RNA binding protein Arid5a. Sci. Signal. 11:eaat4617. 10.1126/scisignal.aat4617 - DOI - PMC - PubMed
-
- Binnewies, M., Roberts E.W., Kersten K., Chan V., Fearon D.F., Merad M., Coussens L.M., Gabrilovich D.I., Ostrand-Rosenberg S., Hedrick C.C., et al. . 2018. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24:541–550. 10.1038/s41591-018s4150014-x - DOI - PMC - PubMed
-
- Buchmann, A., Ruggeri B., Klein-Szanto A.J., and Balmain A.. 1991. Progression of squamous carcinoma cells to spindle carcinomas of mouse skin is associated with an imbalance of H-ras alleles on chromosome 7. Cancer Res. 51:4097–4101. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
