

Clinical characteristics and prognosis of amyotrophic lateral sclerosis with autoimmune diseases
- PMID: 35390090
- PMCID: PMC8989203
- DOI: 10.1371/journal.pone.0266529
Clinical characteristics and prognosis of amyotrophic lateral sclerosis with autoimmune diseases
Abstract
Introduction: The occurrence of autoimmune diseases (AIDs) in amyotrophic lateral sclerosis (ALS) patients is widely reported, but little is known about the associated clinical phenotype. This study aims to evaluate the clinical features and prognosis of ALS patients with AID.
Methods: This retrospective study was based on the ALS Registry dataset of Peking Union Medical College Hospital from 2013 to 2020. Clinical features and inflammatory biomarkers at registration were compared between ALS patients with coexisting AIDs and those without (controls). The medical records of immunotherapy were also collected. The Kaplan-Meier method and Cox proportional hazard model were used to study the survival of ALS patients.
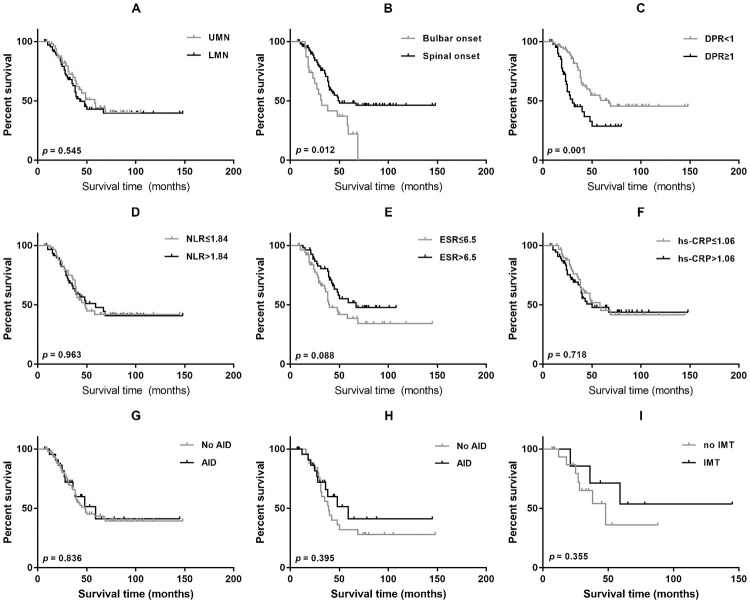
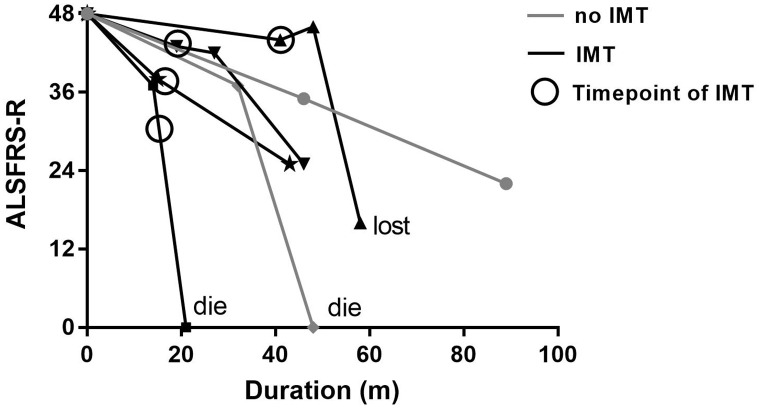
Results: There are 26 (1.6%) ALS patients with AIDs in our database. The ALS patients with AIDs had older ages at onset and poorer respiratory function than controls (p<0.05). After propensity score matching by sex, onset age, and disease duration, the difference in respiratory function remained significant between groups. We found no differences in overall survival between ALS patients with and without AIDs before and after matching (p = 0.836; p = 0.395). Older age at onset, rapid disease progression, and lower erythrocyte sedimentation rate (ESR) were associated with shorter survival (p<0.05). Among ALS patients with AIDs, 8 (30.8%) had a history of immunotherapy and showed slightly prolonged survival compared with those without immunotherapy, but the results did not reach statistical significance (p = 0.355).
Conclusions: Patients with coexisting ALS and AIDs had older onset age and poorer respiratory function but similar overall survival than those with pure ALS.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Natural history and clinical features of sporadic amyotrophic lateral sclerosis in China.J Neurol Neurosurg Psychiatry. 2015 Oct;86(10):1075-81. doi: 10.1136/jnnp-2015-310471. Epub 2015 Jun 29. J Neurol Neurosurg Psychiatry. 2015. PMID: 26124198
-
[The changes of clinical characteristics in 100 Japanese amyotrophic lateral sclerosis patients between 1980 and 2000].Rinsho Shinkeigaku. 2003 Jul;43(7):385-91. Rinsho Shinkeigaku. 2003. PMID: 14582363 Japanese.
-
Survival rate of patients with amyotrophic lateral sclerosis in Wakayama Prefecture, Japan, 1966 to 2005.J Neurol Sci. 2008 May 15;268(1-2):95-101. doi: 10.1016/j.jns.2007.11.011. Epub 2007 Dec 31. J Neurol Sci. 2008. PMID: 18164728
-
A retrospective study of the clinical phenotype and predictors of survival in non-Caucasian Hispanic patients with amyotrophic lateral sclerosis.BMC Neurol. 2019 Oct 29;19(1):261. doi: 10.1186/s12883-019-1459-3. BMC Neurol. 2019. PMID: 31664949 Free PMC article.
-
Slower disease progression and prolonged survival in contemporary patients with amyotrophic lateral sclerosis: is the natural history of amyotrophic lateral sclerosis changing?Arch Neurol. 2006 Aug;63(8):1139-43. doi: 10.1001/archneur.63.8.1139. Arch Neurol. 2006. PMID: 16908741
Cited by
-
Comprehensive analysis of autoimmune-related genes in amyotrophic lateral sclerosis from the perspective of 3P medicine.EPMA J. 2022 Oct 12;13(4):699-723. doi: 10.1007/s13167-022-00299-w. eCollection 2022 Dec. EPMA J. 2022. PMID: 36505891 Free PMC article.
-
Prevalence and impact of comorbidities in amyotrophic lateral sclerosis.J Neural Transm (Vienna). 2025 Jun 14. doi: 10.1007/s00702-025-02971-7. Online ahead of print. J Neural Transm (Vienna). 2025. PMID: 40515812 Review.
-
Rare Diseases, Spotlighting Amyotrophic Lateral Sclerosis, Huntington's Disease, and Myasthenia Gravis: Insights from Landscape Analysis of Current Research.Biochemistry. 2025 Apr 15;64(8):1698-1719. doi: 10.1021/acs.biochem.4c00722. Epub 2025 Apr 1. Biochemistry. 2025. PMID: 40169538 Free PMC article. Review.
-
Immune-mediated diseases are associated with a higher risk of ALS incidence: a prospective cohort study from the UK Biobank.Front Immunol. 2024 Mar 5;15:1356132. doi: 10.3389/fimmu.2024.1356132. eCollection 2024. Front Immunol. 2024. PMID: 38504981 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
