

Phenotype-driven approaches to enhance variant prioritization and diagnosis of rare disease
- PMID: 35391505
- PMCID: PMC9288531
- DOI: 10.1002/humu.24380
Phenotype-driven approaches to enhance variant prioritization and diagnosis of rare disease
Abstract
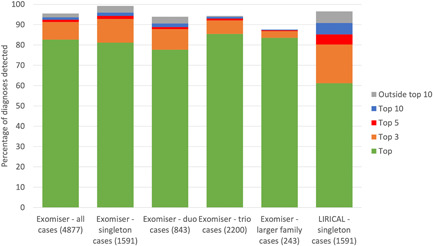
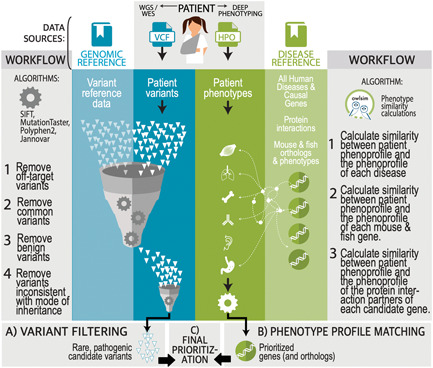
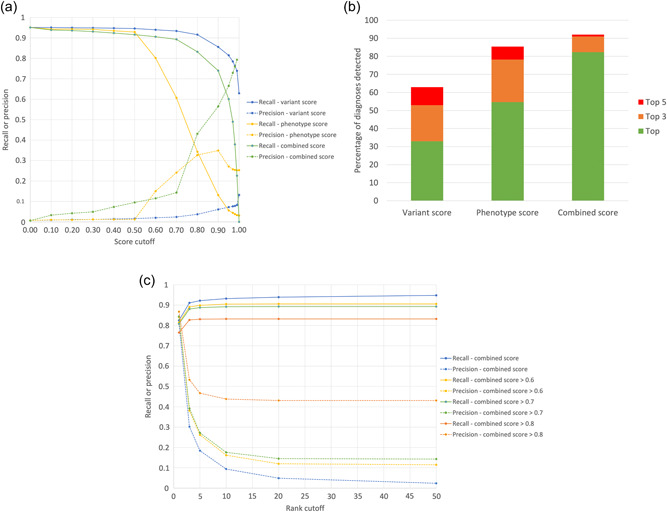
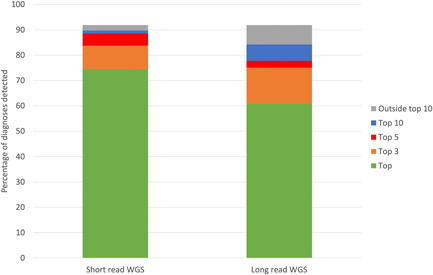
Rare disease diagnostics and disease gene discovery have been revolutionized by whole-exome and genome sequencing but identifying the causative variant(s) from the millions in each individual remains challenging. The use of deep phenotyping of patients and reference genotype-phenotype knowledge, alongside variant data such as allele frequency, segregation, and predicted pathogenicity, has proved an effective strategy to tackle this issue. Here we review the numerous tools that have been developed to automate this approach and demonstrate the power of such an approach on several thousand diagnosed cases from the 100,000 Genomes Project. Finally, we discuss the challenges that need to be overcome if we are going to improve detection rates and help the majority of patients that still remain without a molecular diagnosis after state-of-the-art genomic interpretation.
Keywords: diagnostics; phenotypes; rare disease; variant prioritization.
© 2022 The Authors. Human Mutation published by Wiley Periodicals LLC.
Conflict of interest statement
Julius Jacobsen and Damian Smedley declare they previously acted as part‐time consultants for Congenica Ltd. The other authors declare no other potential conflicts of interest.
Figures
References
-
- Antanaviciute, A. , Watson, C. M. , Harrison, S. M. , Lascelles, C. , Crinnion, L. , Markham, A. F. , Bonthron, D. T. , & Carr, I. M. (2015). OVA: Integrating molecular and physical phenotype data from multiple biomedical domain ontologies with variant filtering for enhanced variant prioritization. Bioinformatics, 31, 3822–3829. - PMC - PubMed
