

Mitophagy and Neurodegeneration: Between the Knowns and the Unknowns
- PMID: 35392168
- PMCID: PMC8981085
- DOI: 10.3389/fcell.2022.837337
Mitophagy and Neurodegeneration: Between the Knowns and the Unknowns
Abstract
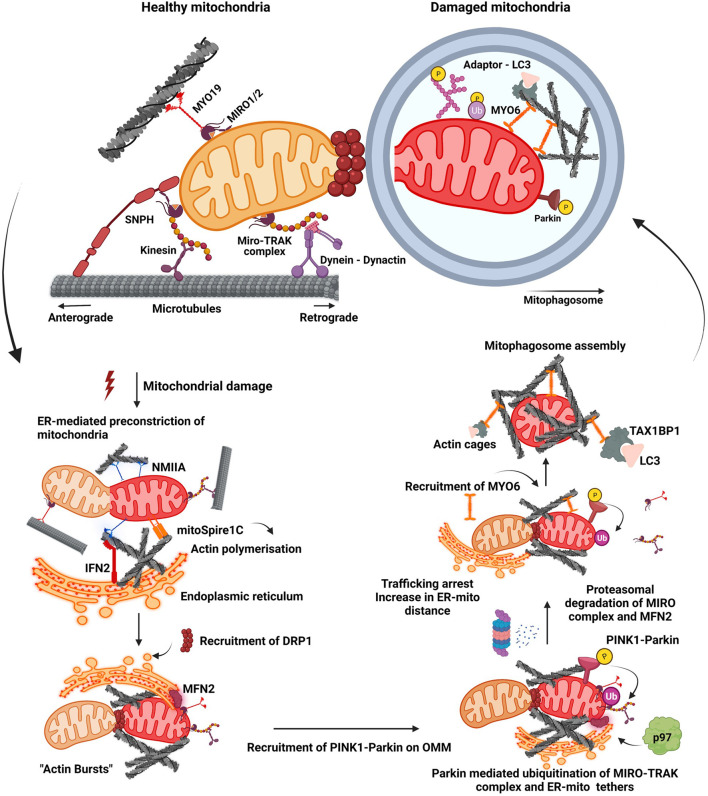
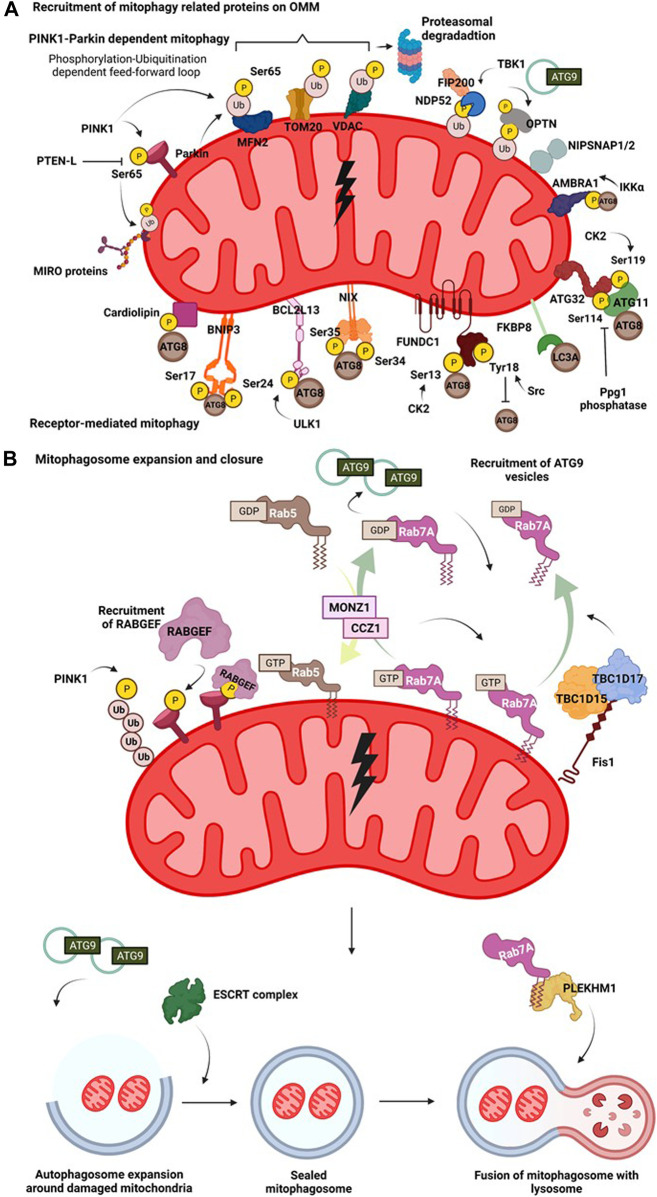
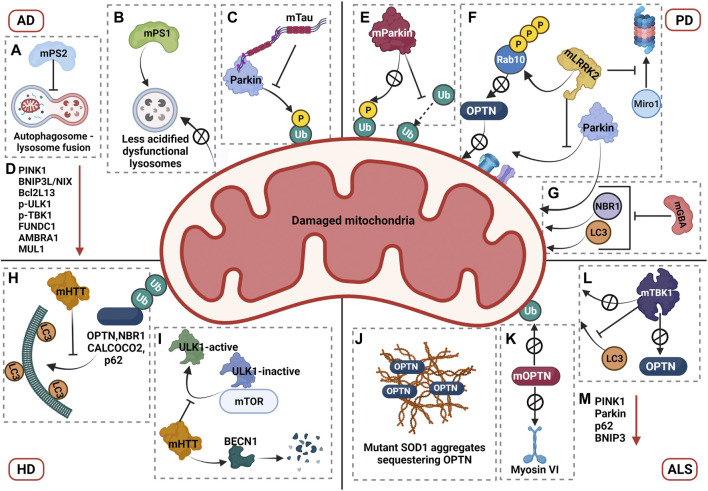
Macroautophagy (henceforth autophagy) an evolutionary conserved intracellular pathway, involves lysosomal degradation of damaged and superfluous cytosolic contents to maintain cellular homeostasis. While autophagy was initially perceived as a bulk degradation process, a surfeit of studies in the last 2 decades has revealed that it can also be selective in choosing intracellular constituents for degradation. In addition to the core autophagy machinery, these selective autophagy pathways comprise of distinct molecular players that are involved in the capture of specific cargoes. The diverse organelles that are degraded by selective autophagy pathways are endoplasmic reticulum (ERphagy), lysosomes (lysophagy), mitochondria (mitophagy), Golgi apparatus (Golgiphagy), peroxisomes (pexophagy) and nucleus (nucleophagy). Among these, the main focus of this review is on the selective autophagic pathway involved in mitochondrial turnover called mitophagy. The mitophagy pathway encompasses diverse mechanisms involving a complex interplay of a multitude of proteins that confers the selective recognition of damaged mitochondria and their targeting to degradation via autophagy. Mitophagy is triggered by cues that signal the mitochondrial damage such as disturbances in mitochondrial fission-fusion dynamics, mitochondrial membrane depolarisation, enhanced ROS production, mtDNA damage as well as developmental cues such as erythrocyte maturation, removal of paternal mitochondria, cardiomyocyte maturation and somatic cell reprogramming. As research on the mechanistic aspects of this complex pathway is progressing, emerging roles of new players such as the NIPSNAP proteins, Miro proteins and ER-Mitochondria contact sites (ERMES) are being explored. Although diverse aspects of this pathway are being investigated in depth, several outstanding questions such as distinct molecular players of basal mitophagy, selective dominance of a particular mitophagy adapter protein over the other in a given physiological condition, molecular mechanism of how specific disease mutations affect this pathway remain to be addressed. In this review, we aim to give an overview with special emphasis on molecular and signalling pathways of mitophagy and its dysregulation in neurodegenerative disorders.
Keywords: mitochondrial dynamics; mitochondrial dysfunction; mitophagy; neurodegenaration; phosphorylation; ubiquitination.
Copyright © 2022 Jetto, Nambiar and Manjithaya.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials