

Dissecting the clinical heterogeneity of early-onset Alzheimer's disease
- PMID: 35393555
- PMCID: PMC9156414
- DOI: 10.1038/s41380-022-01531-9
Dissecting the clinical heterogeneity of early-onset Alzheimer's disease
Abstract
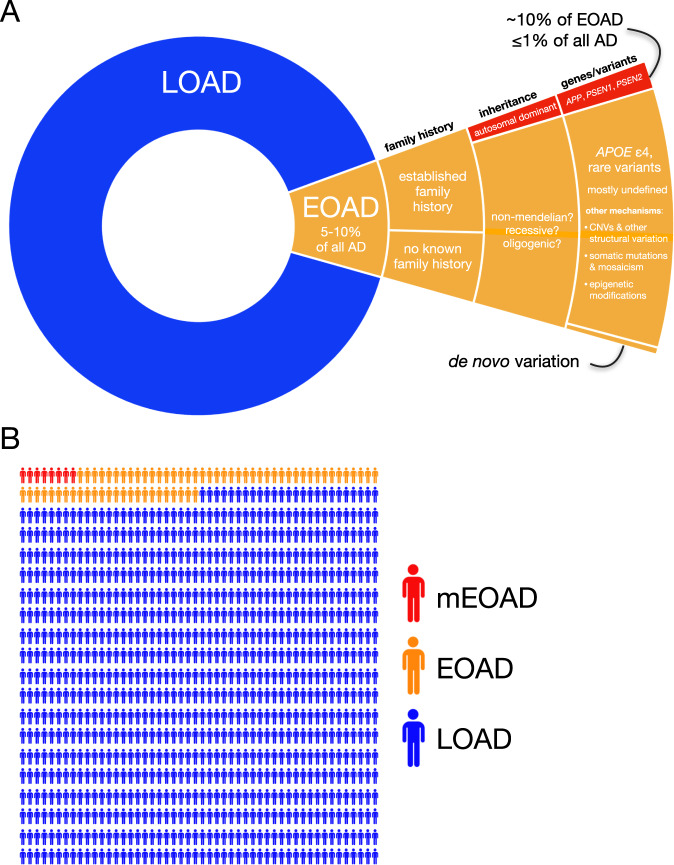
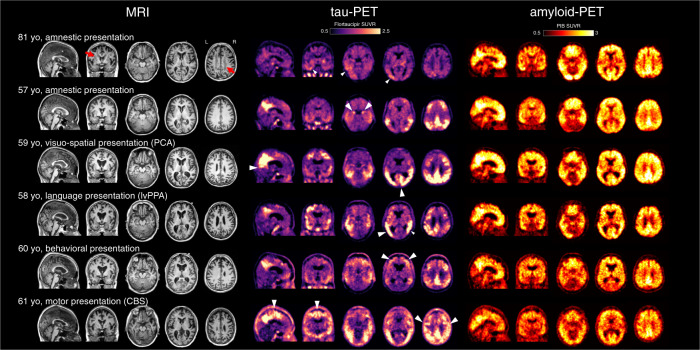
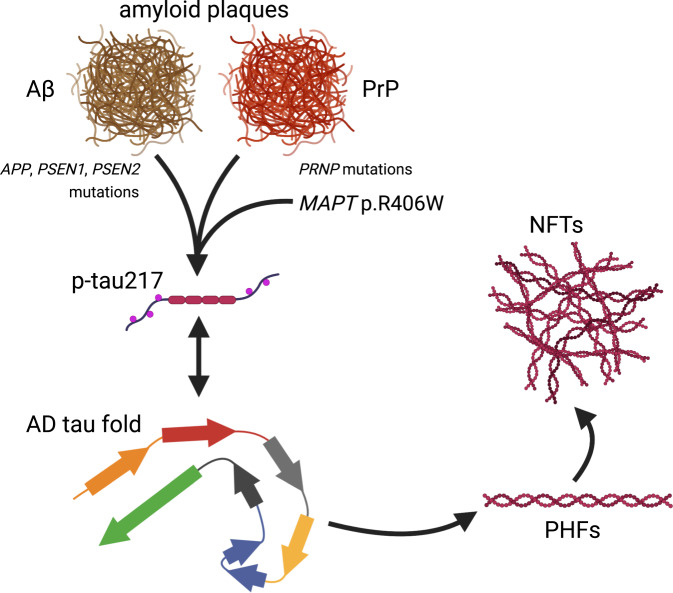
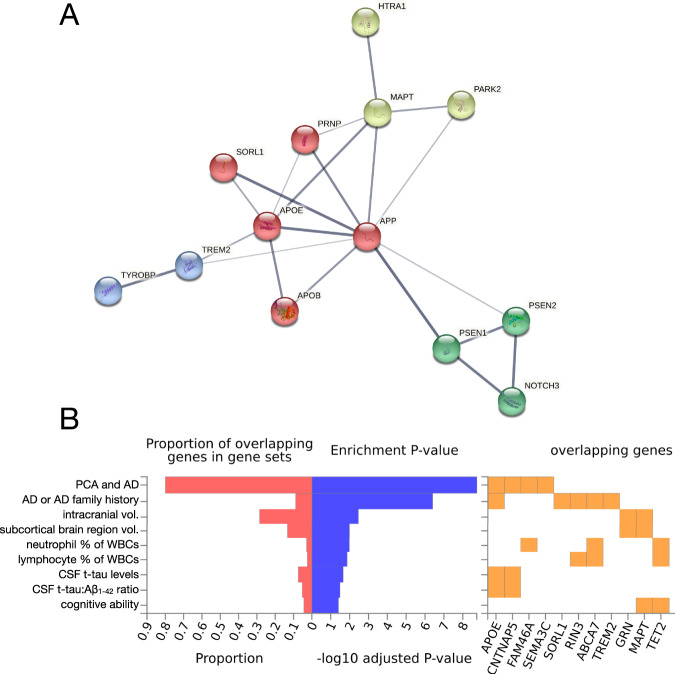
Early-onset Alzheimer's disease (EOAD) is a rare but particularly devastating form of AD. Though notable for its high degree of clinical heterogeneity, EOAD is defined by the same neuropathological hallmarks underlying the more common, late-onset form of AD. In this review, we describe the various clinical syndromes associated with EOAD, including the typical amnestic phenotype as well as atypical variants affecting visuospatial, language, executive, behavioral, and motor functions. We go on to highlight advances in fluid biomarker research and describe how molecular, structural, and functional neuroimaging can be used not only to improve EOAD diagnostic acumen but also enhance our understanding of fundamental pathobiological changes occurring years (and even decades) before the onset of symptoms. In addition, we discuss genetic variation underlying EOAD, including pathogenic variants responsible for the well-known mendelian forms of EOAD as well as variants that may increase risk for the much more common forms of EOAD that are either considered to be sporadic or lack a clear autosomal-dominant inheritance pattern. Intriguingly, specific pathogenic variants in PRNP and MAPT-genes which are more commonly associated with other neurodegenerative diseases-may provide unexpectedly important insights into the formation of AD tau pathology. Genetic analysis of the atypical clinical syndromes associated with EOAD will continue to be challenging given their rarity, but integration of fluid biomarker data, multimodal imaging, and various 'omics techniques and their application to the study of large, multicenter cohorts will enable future discoveries of fundamental mechanisms underlying the development of EOAD and its varied clinical presentations.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
