

Germline variants in tumor suppressor FBXW7 lead to impaired ubiquitination and a neurodevelopmental syndrome
- PMID: 35395208
- PMCID: PMC9069070
- DOI: 10.1016/j.ajhg.2022.03.002
Germline variants in tumor suppressor FBXW7 lead to impaired ubiquitination and a neurodevelopmental syndrome
Abstract
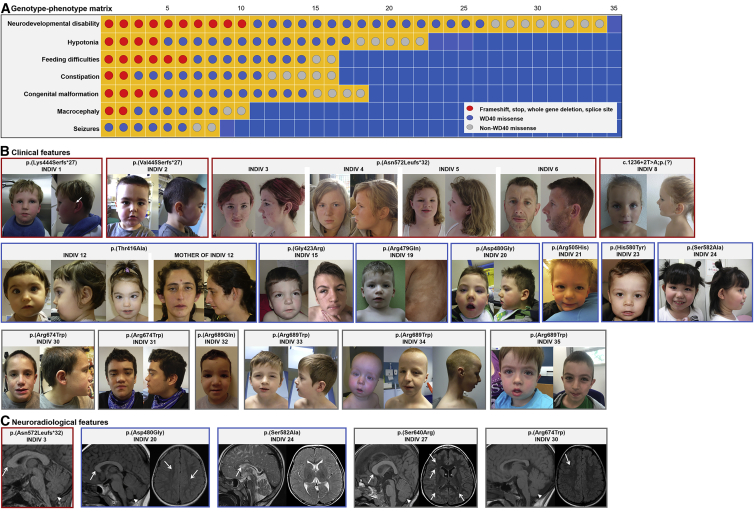
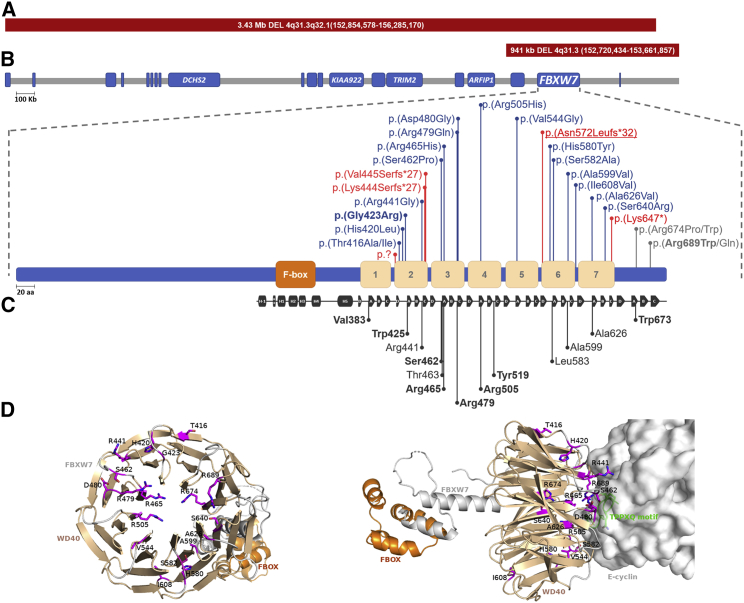
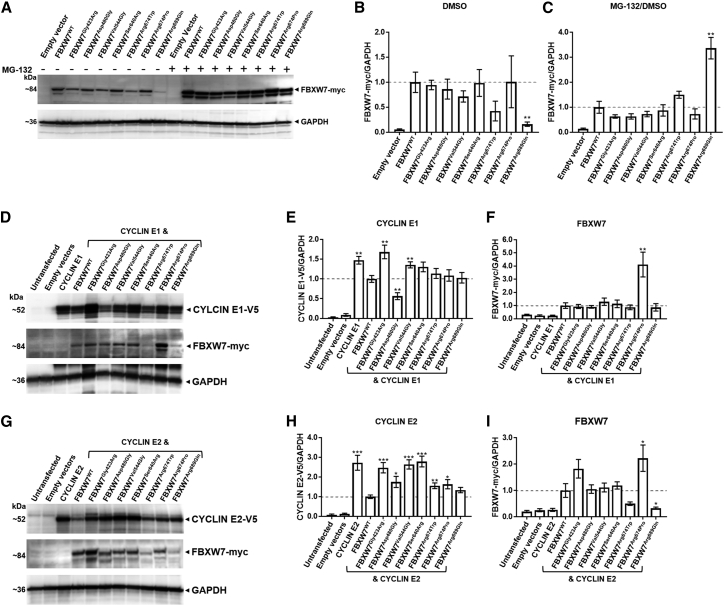
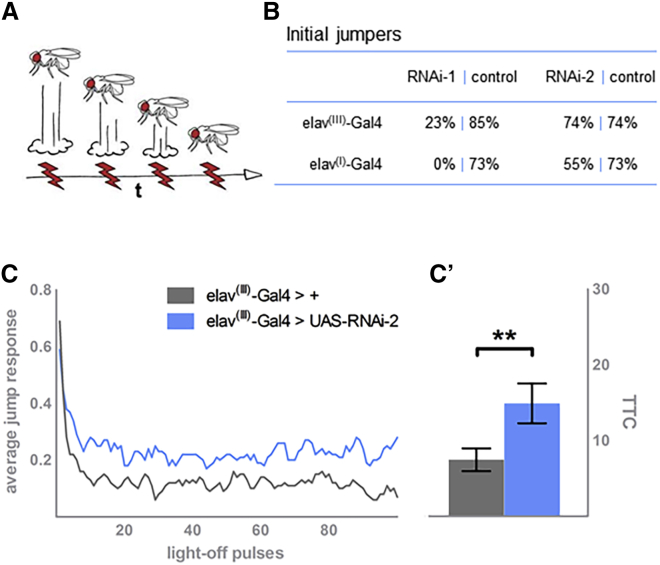
Neurodevelopmental disorders are highly heterogenous conditions resulting from abnormalities of brain architecture and/or function. FBXW7 (F-box and WD-repeat-domain-containing 7), a recognized developmental regulator and tumor suppressor, has been shown to regulate cell-cycle progression and cell growth and survival by targeting substrates including CYCLIN E1/2 and NOTCH for degradation via the ubiquitin proteasome system. We used a genotype-first approach and global data-sharing platforms to identify 35 individuals harboring de novo and inherited FBXW7 germline monoallelic chromosomal deletions and nonsense, frameshift, splice-site, and missense variants associated with a neurodevelopmental syndrome. The FBXW7 neurodevelopmental syndrome is distinguished by global developmental delay, borderline to severe intellectual disability, hypotonia, and gastrointestinal issues. Brain imaging detailed variable underlying structural abnormalities affecting the cerebellum, corpus collosum, and white matter. A crystal-structure model of FBXW7 predicted that missense variants were clustered at the substrate-binding surface of the WD40 domain and that these might reduce FBXW7 substrate binding affinity. Expression of recombinant FBXW7 missense variants in cultured cells demonstrated impaired CYCLIN E1 and CYCLIN E2 turnover. Pan-neuronal knockdown of the Drosophila ortholog, archipelago, impaired learning and neuronal function. Collectively, the data presented herein provide compelling evidence of an F-Box protein-related, phenotypically variable neurodevelopmental disorder associated with monoallelic variants in FBXW7.
Keywords: F-box protein; FBXW7; Neurodevelopment; brain malformation; epilepsy; gastrointestinal issues; global developmental delay; hypotonia; intellectual disability; macrocephaly.
Copyright © 2022 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests I.E.S. has served on scientific advisory boards for UCB, Eisai, GlaxoSmithKline, BioMarin, Nutricia, Rogcon, Chiesi, Encoded Therapeutics, Xenon Pharmaceuticals, and Knopp Biosciences; has received speaker honoraria from GlaxoSmithKline, UCB, BioMarin, Biocodex, and Eisai; has received funding for travel from UCB, Biocodex, GlaxoSmithKline, Biomarin and Eisai; has served as an investigator for Zogenix, Zynerba, Ultragenyx, GW Pharma, UCB, Eisai, Anavex Life Sciences, Ovid Therapeutics, Epygenyx, Encoded Therapeutics and Marinus; and has consulted for Zynerba Pharmaceuticals, Atheneum Partners, Ovid Therapeutics, Care Beyond Diagnosis, Epilepsy Consortium and UCB. She may accrue future revenue on pending patent WO2009/086591; her patent for SCN1A testing is held by Bionomics and is licensed to various diagnostic companies; and she has a patent for a molecular diagnostic/therapeutic target for benign familial infantile epilepsy (BFIE) (PRRT2), WO/2013/059884. She receives and/or has received research support from the National Health and Medical Research Council of Australia, Medical Research Future Fund, Health Research Council of New Zealand, CURE, Australian Epilepsy Research Fund, and the National Institute of Neurological Disorders and Stroke of the National Institutes of Health. J.P. is co-chief scientific officer for Global Gene Corp. All other authors declare no competing interests.
Figures
References
-
- Bruel A.L., Vitobello A., Tran Mau-Them F., Nambot S., Sorlin A., Denommé-Pichon A.S., Delanne J., Moutton S., Callier P., Duffourd Y., et al. Next-generation sequencing approaches and challenges in the diagnosis of developmental anomalies and intellectual disability. Clin. Genet. 2020;98:433–444. doi: 10.1111/cge.13764. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
