

Of the many cellular responses activated by TP53, which ones are critical for tumour suppression?
- PMID: 35396345
- PMCID: PMC9090748
- DOI: 10.1038/s41418-022-00996-z
Of the many cellular responses activated by TP53, which ones are critical for tumour suppression?
Abstract
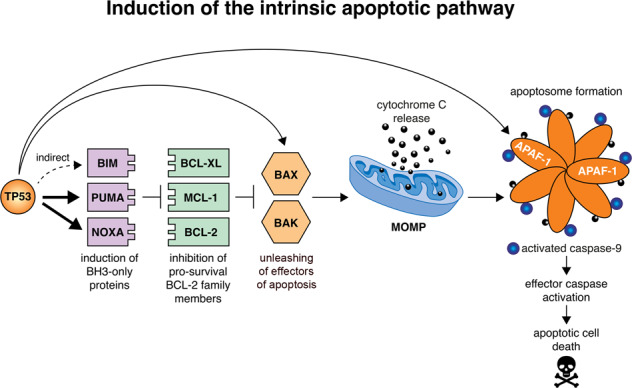
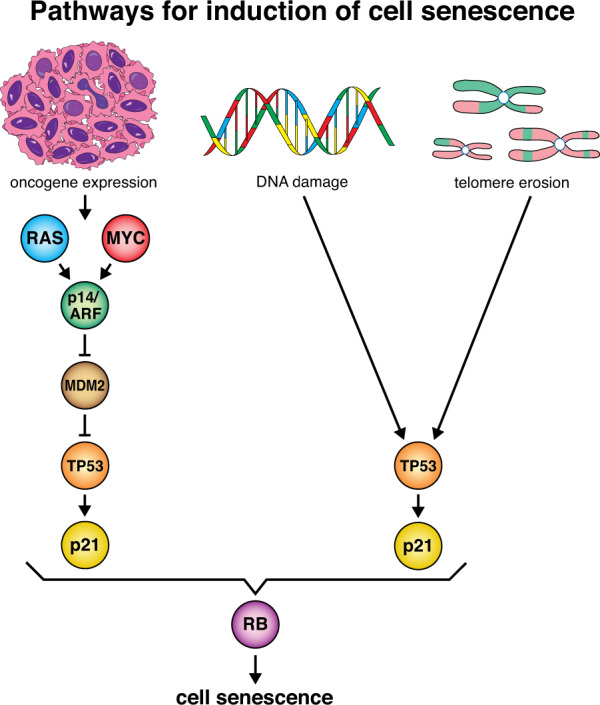
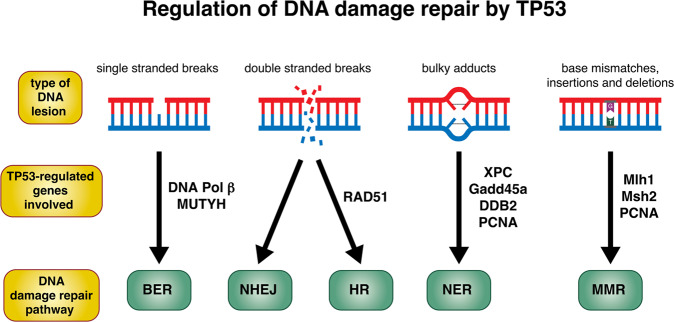
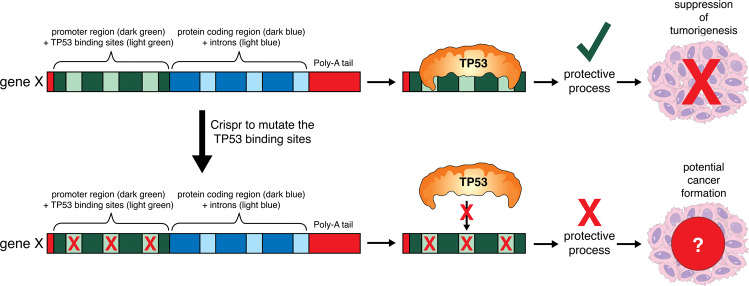
The tumour suppressor TP53 is a master regulator of several cellular processes that collectively suppress tumorigenesis. The TP53 gene is mutated in ~50% of human cancers and these defects usually confer poor responses to therapy. The TP53 protein functions as a homo-tetrameric transcription factor, directly regulating the expression of ~500 target genes, some of them involved in cell death, cell cycling, cell senescence, DNA repair and metabolism. Originally, it was thought that the induction of apoptotic cell death was the principal mechanism by which TP53 prevents the development of tumours. However, gene targeted mice lacking the critical effectors of TP53-induced apoptosis (PUMA and NOXA) do not spontaneously develop tumours. Indeed, even mice lacking the critical mediators for TP53-induced apoptosis, G1/S cell cycle arrest and cell senescence, namely PUMA, NOXA and p21, do not spontaneously develop tumours. This suggests that TP53 must activate additional cellular responses to mediate tumour suppression. In this review, we will discuss the processes by which TP53 regulates cell death, cell cycling/cell senescence, DNA damage repair and metabolic adaptation, and place this in context of current understanding of TP53-mediated tumour suppression.
© 2022. The Author(s), under exclusive licence to ADMC Associazione Differenziamento e Morte Cellulare.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
