Critical Assessment of Metagenome Interpretation: the second round of challenges
- PMID: 35396482
- PMCID: PMC9007738
- DOI: 10.1038/s41592-022-01431-4
Critical Assessment of Metagenome Interpretation: the second round of challenges
Abstract
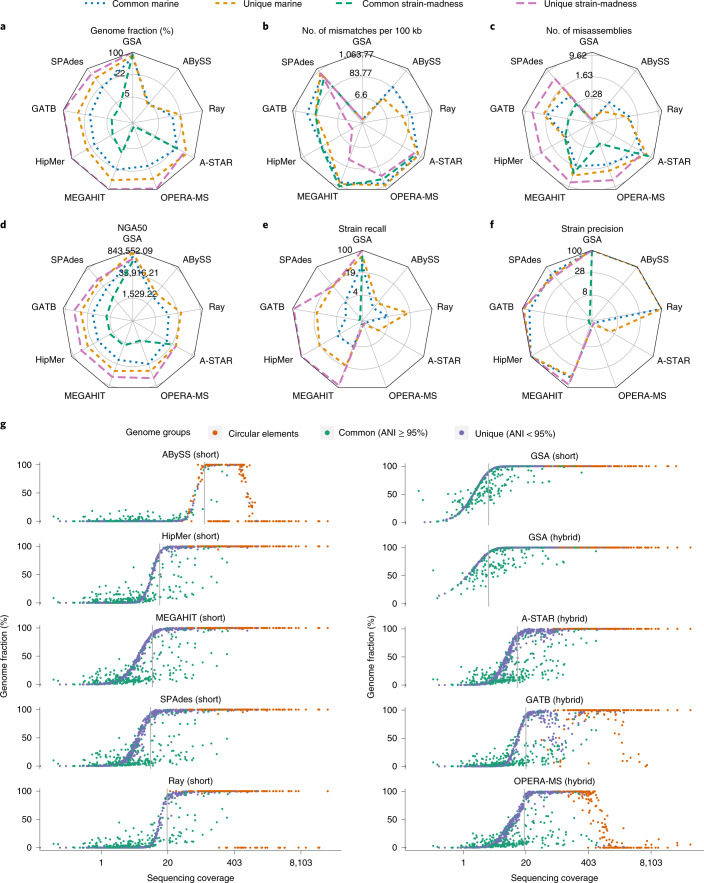
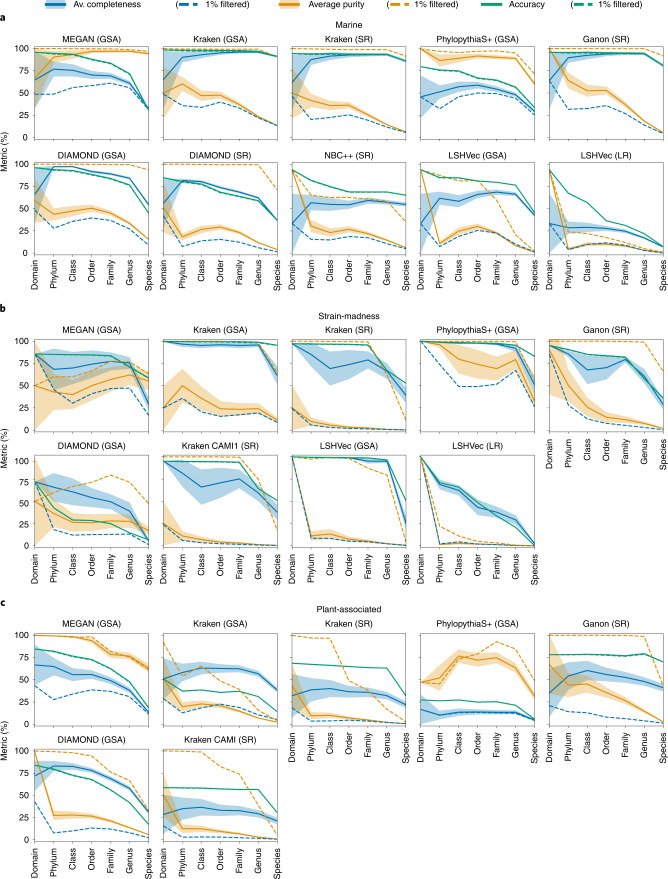
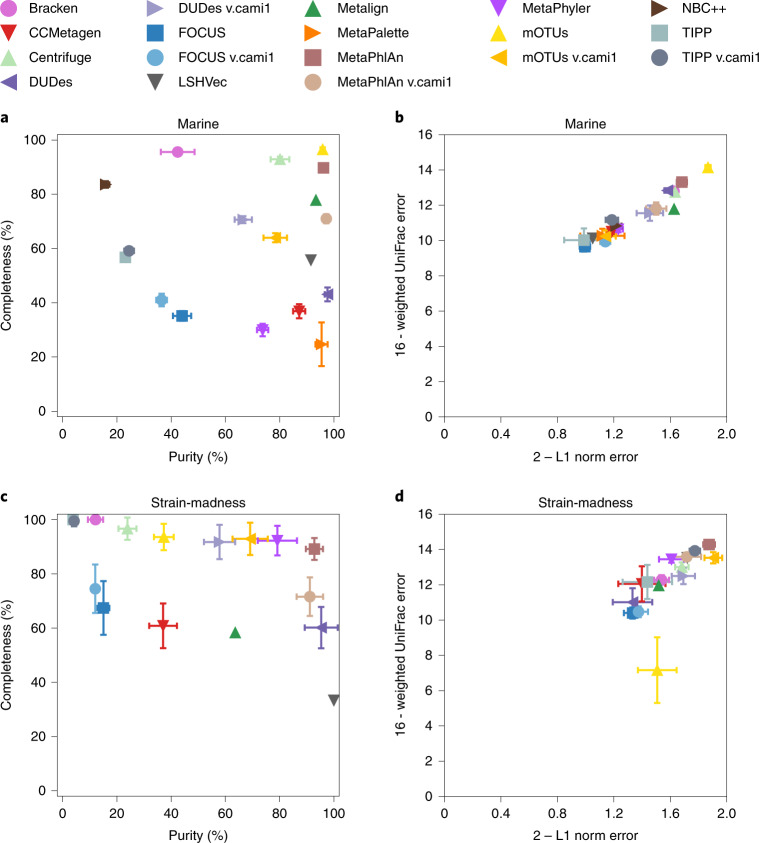
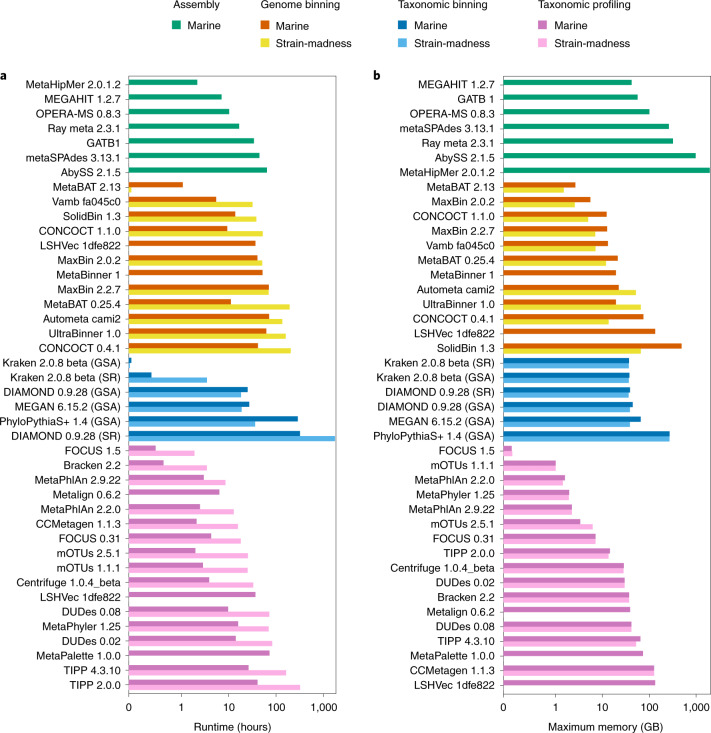
Evaluating metagenomic software is key for optimizing metagenome interpretation and focus of the Initiative for the Critical Assessment of Metagenome Interpretation (CAMI). The CAMI II challenge engaged the community to assess methods on realistic and complex datasets with long- and short-read sequences, created computationally from around 1,700 new and known genomes, as well as 600 new plasmids and viruses. Here we analyze 5,002 results by 76 program versions. Substantial improvements were seen in assembly, some due to long-read data. Related strains still were challenging for assembly and genome recovery through binning, as was assembly quality for the latter. Profilers markedly matured, with taxon profilers and binners excelling at higher bacterial ranks, but underperforming for viruses and Archaea. Clinical pathogen detection results revealed a need to improve reproducibility. Runtime and memory usage analyses identified efficient programs, including top performers with other metrics. The results identify challenges and guide researchers in selecting methods for analyses.
© 2022. The Author(s).
Conflict of interest statement
A.E.D. cofounded Longas Technologies Pty Ltd, a company aimed at development of synthetic long-read sequencing technologies, and is employed by Illumina Australia Pty Ltd. A.C. is employed by Google LLC. L.I. is employed by 10X Genomics. E.R.R. conducted an internship at Empress Therapeutics. E. Georganas is employed by Intel Corporation. G.U. is employed by Amazon.com, Inc. The remaining authors declare no competing interests.
Figures