

DiMeLo-seq: a long-read, single-molecule method for mapping protein-DNA interactions genome wide
- PMID: 35396487
- PMCID: PMC9189060
- DOI: 10.1038/s41592-022-01475-6
DiMeLo-seq: a long-read, single-molecule method for mapping protein-DNA interactions genome wide
Abstract
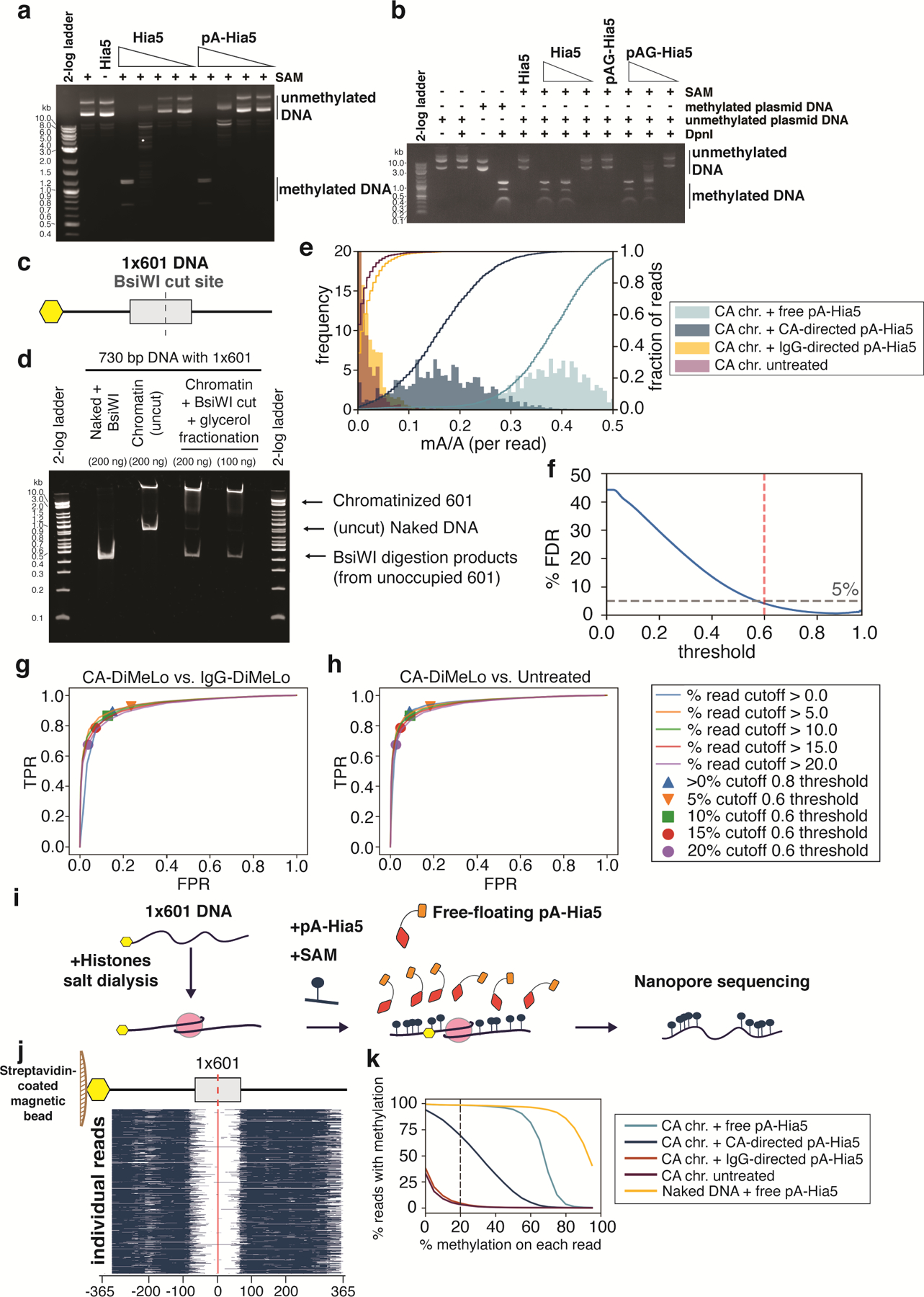
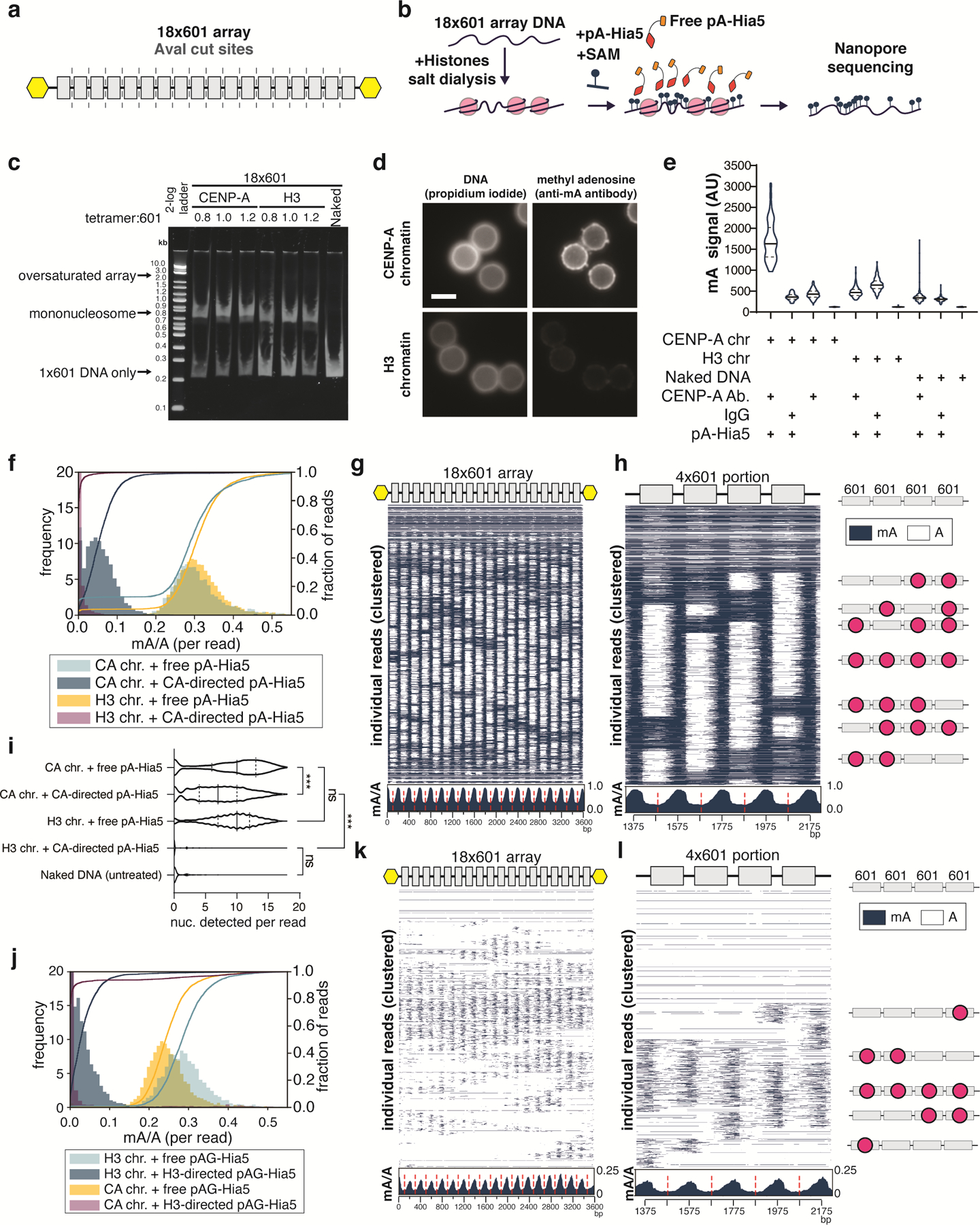
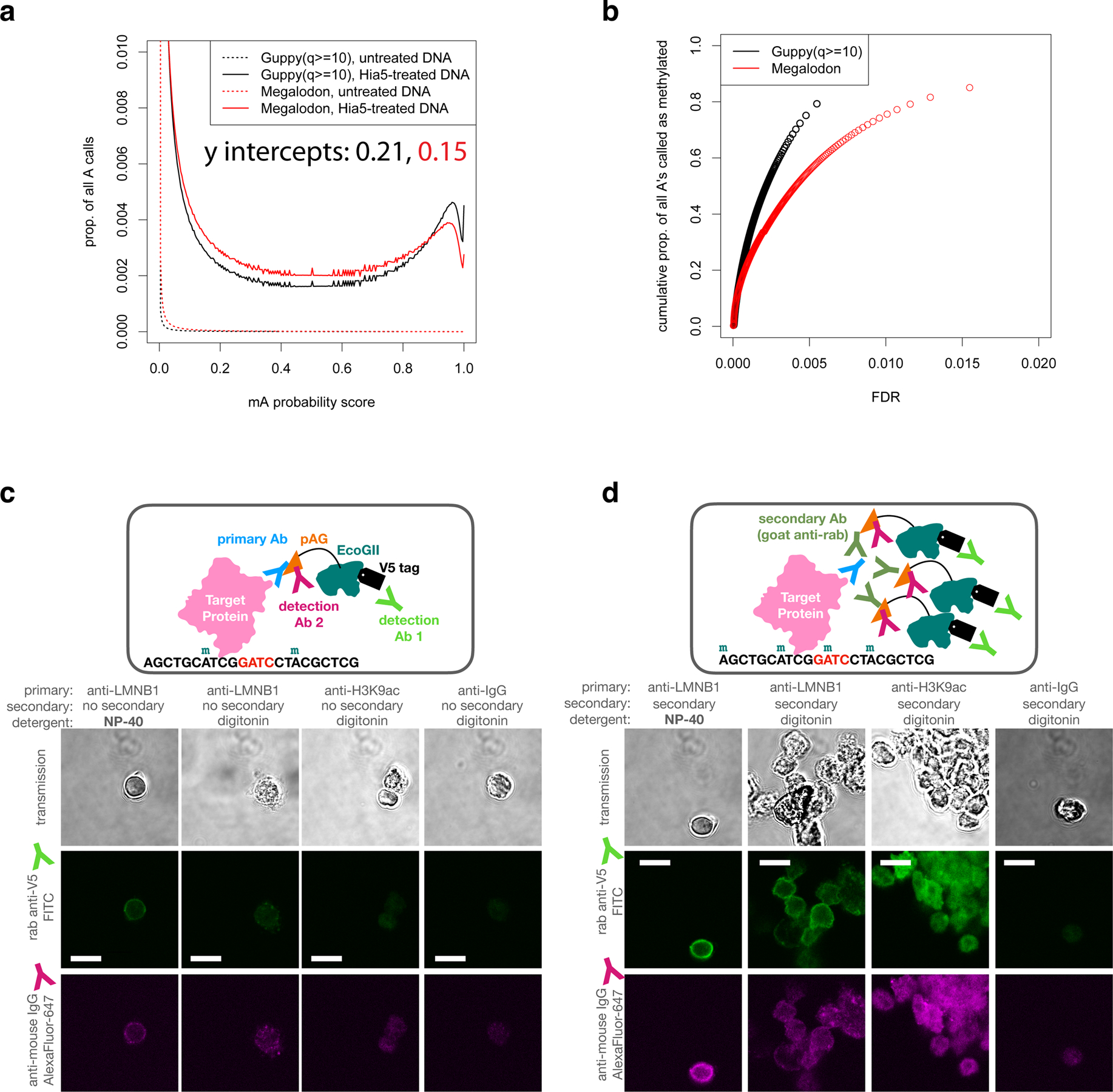
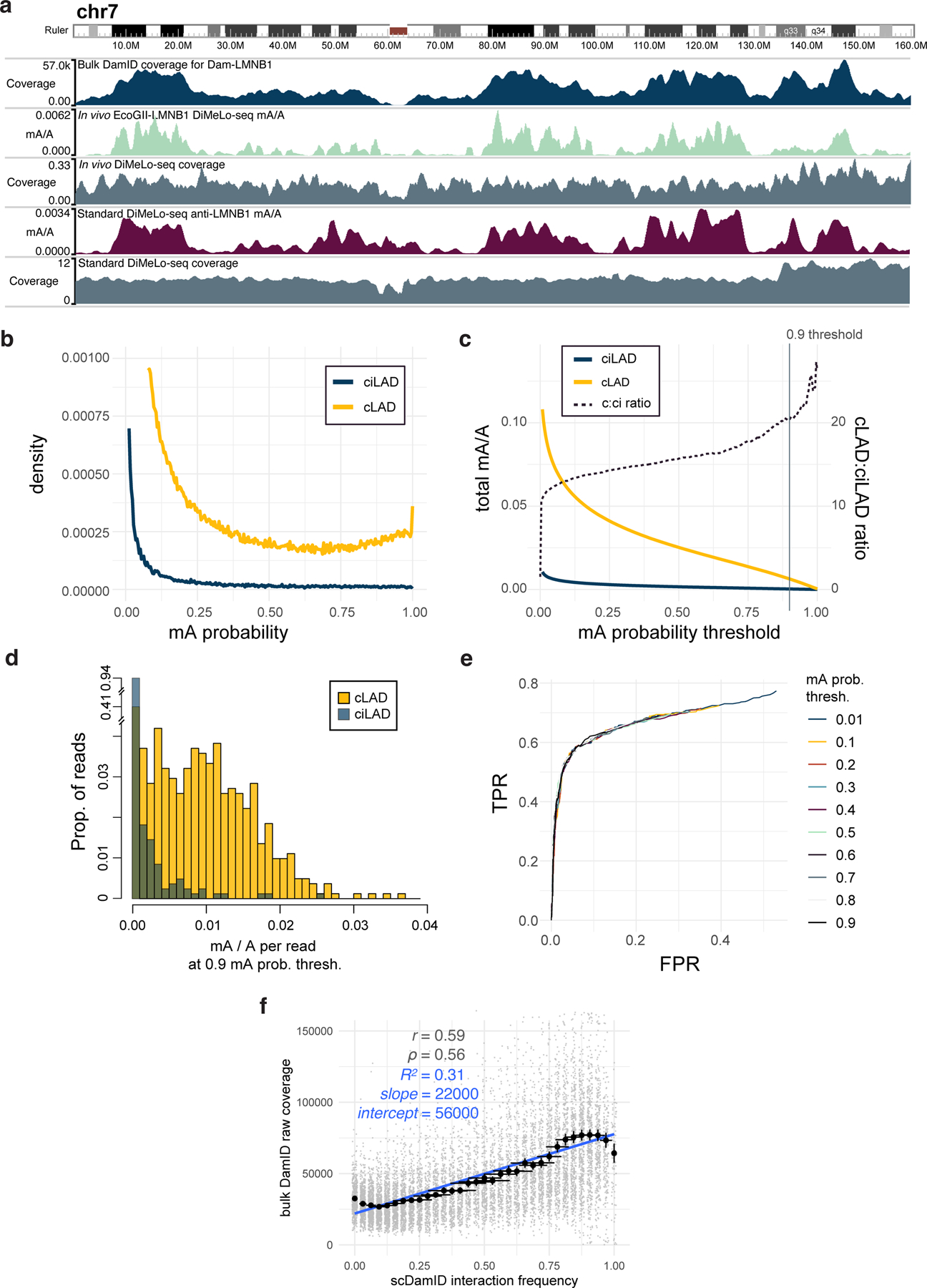
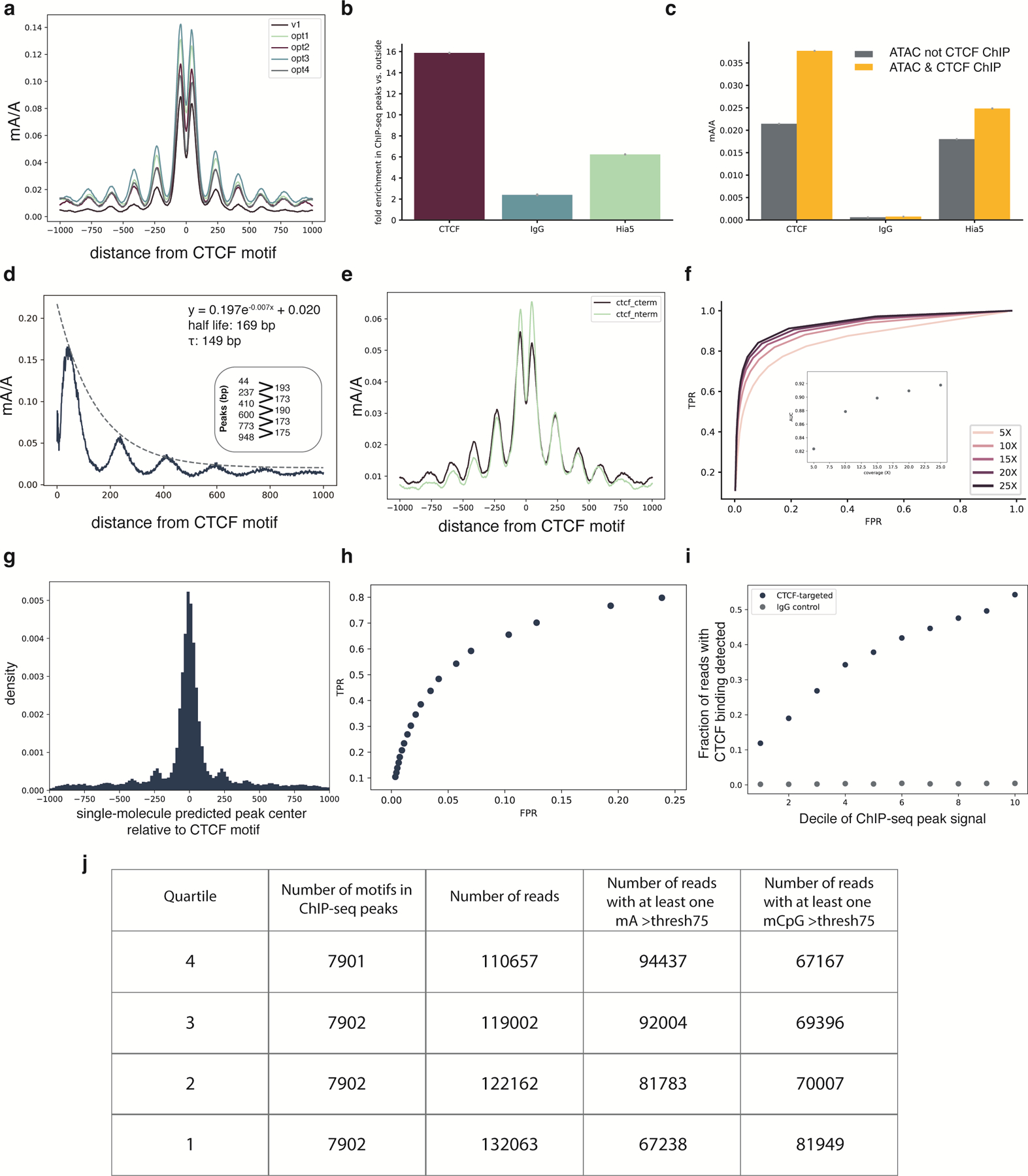
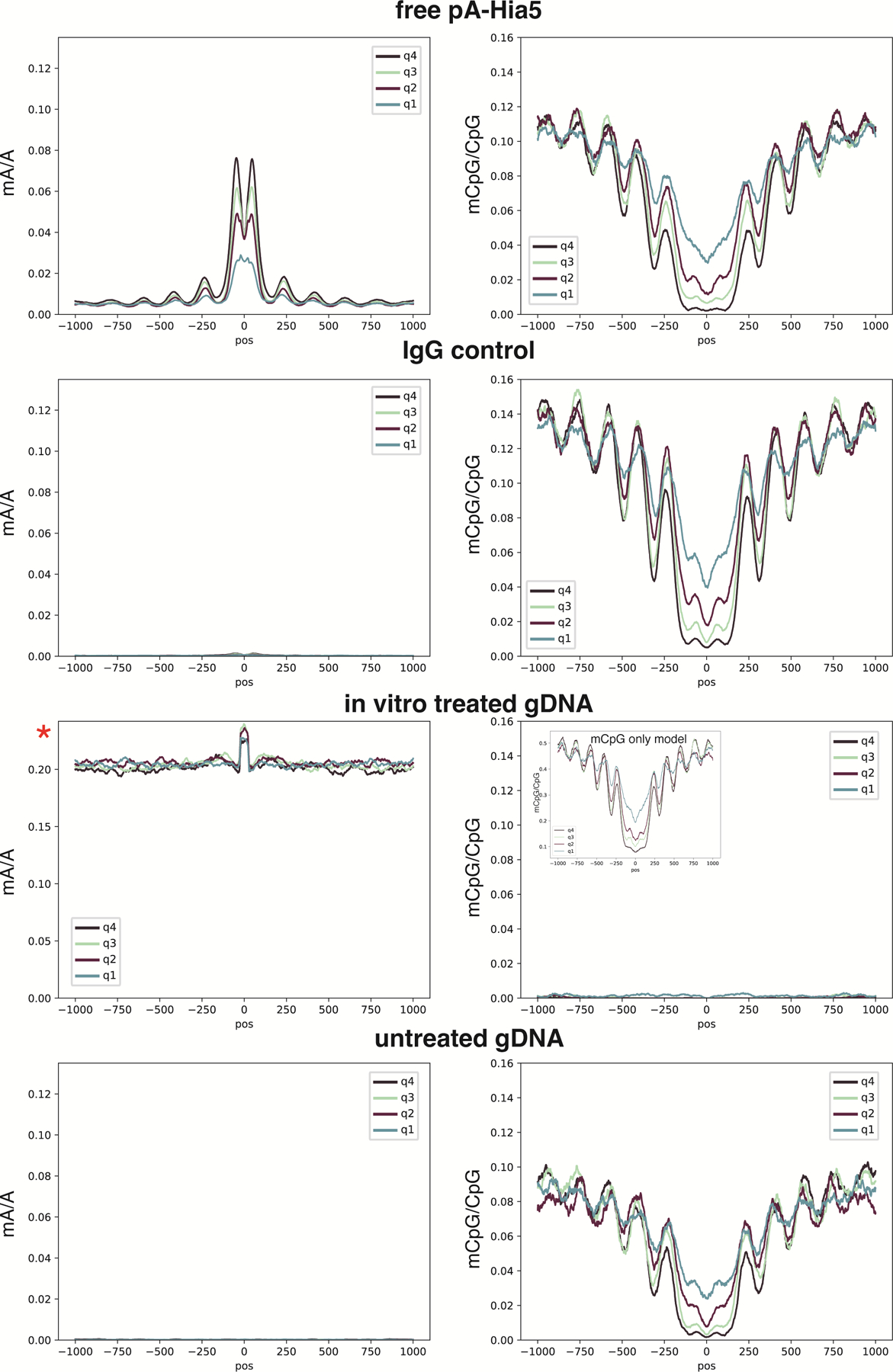
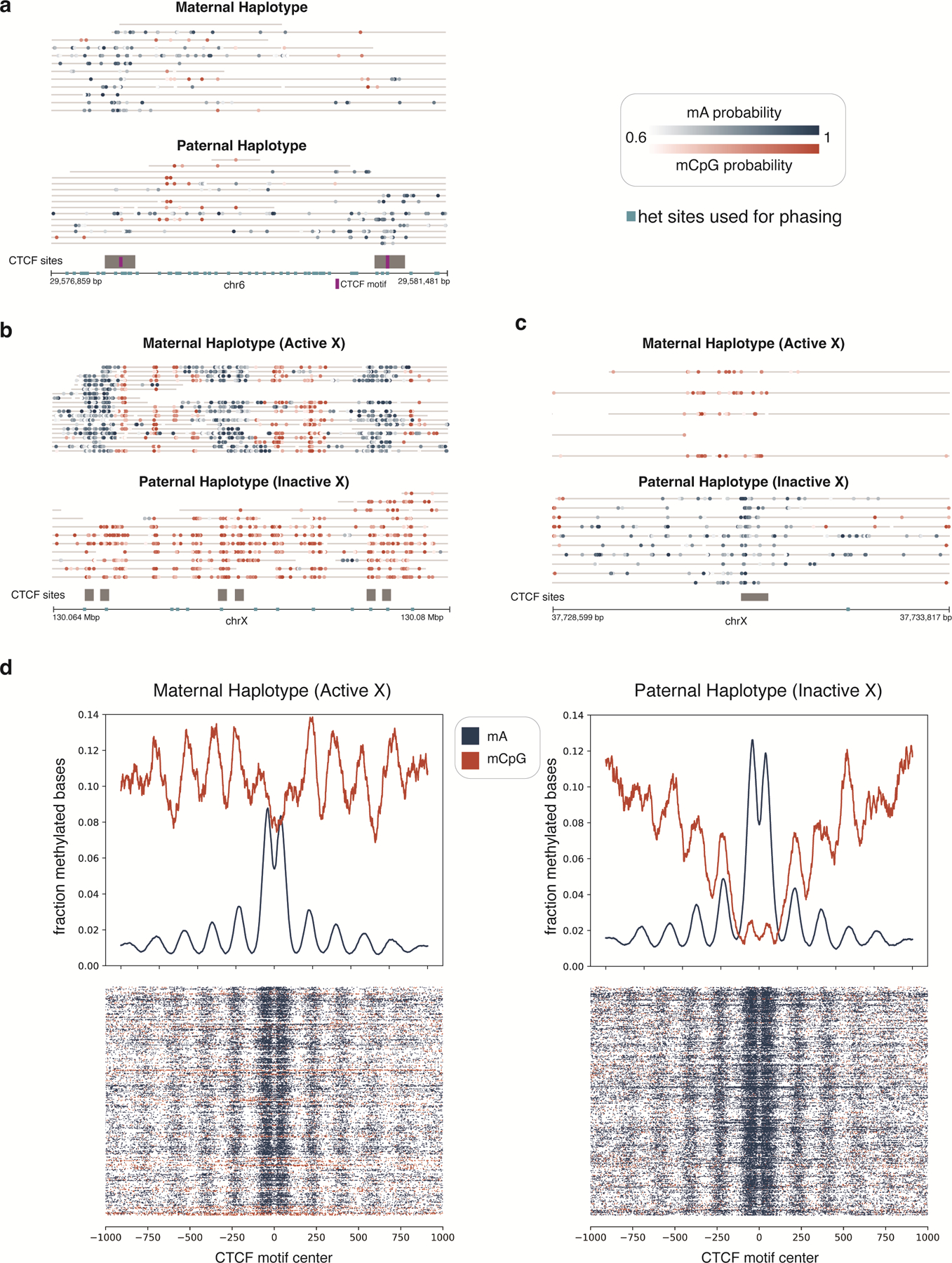
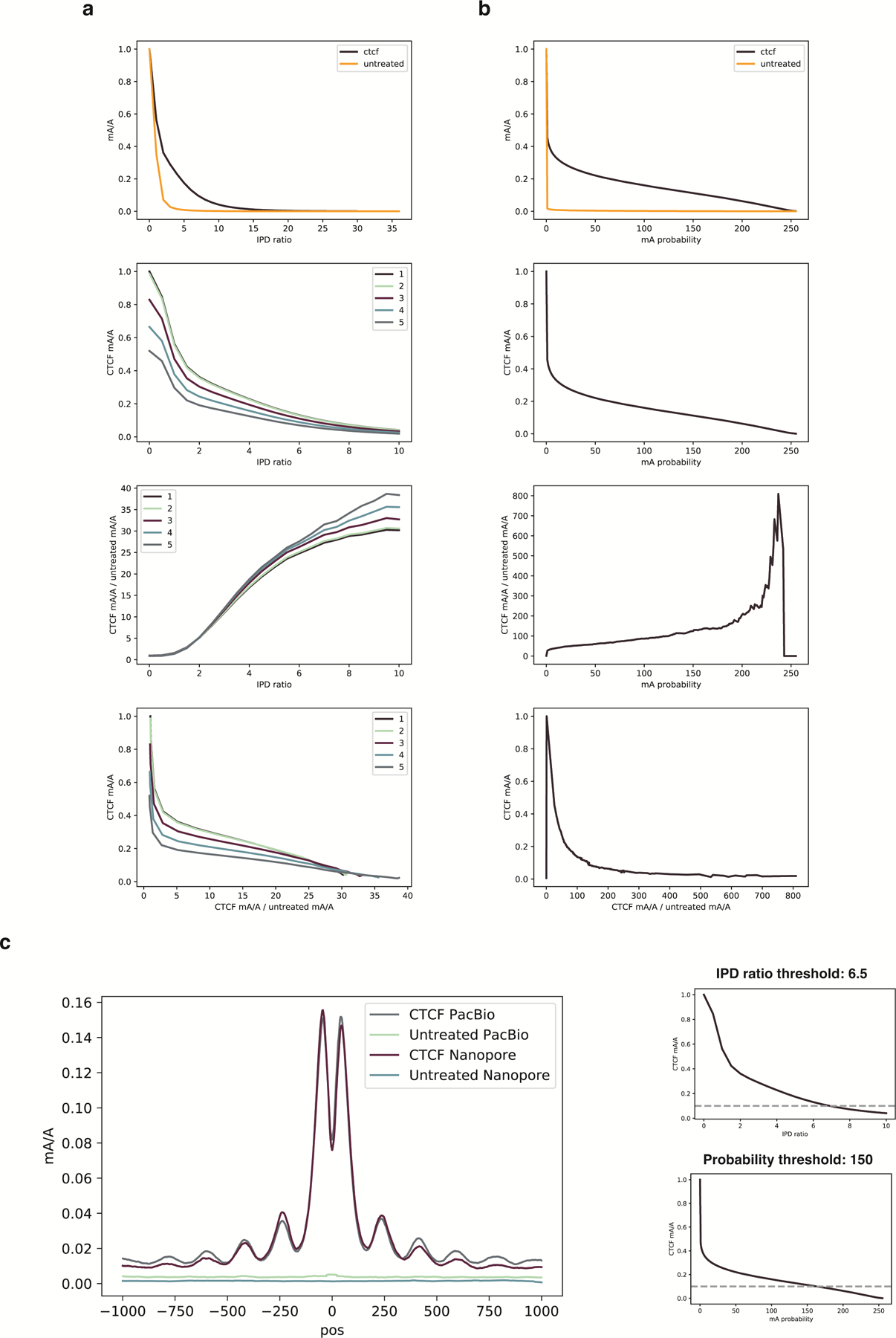
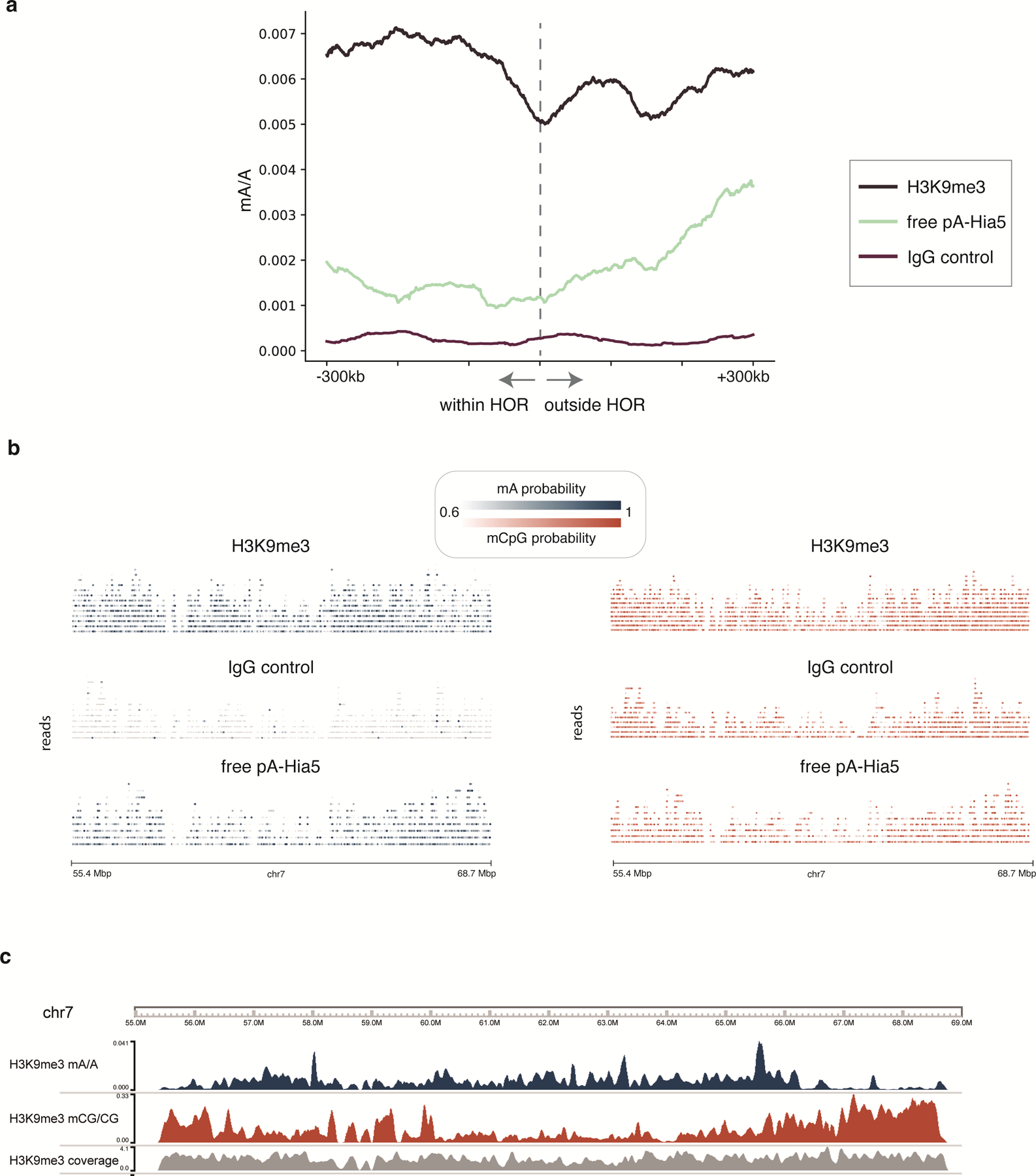
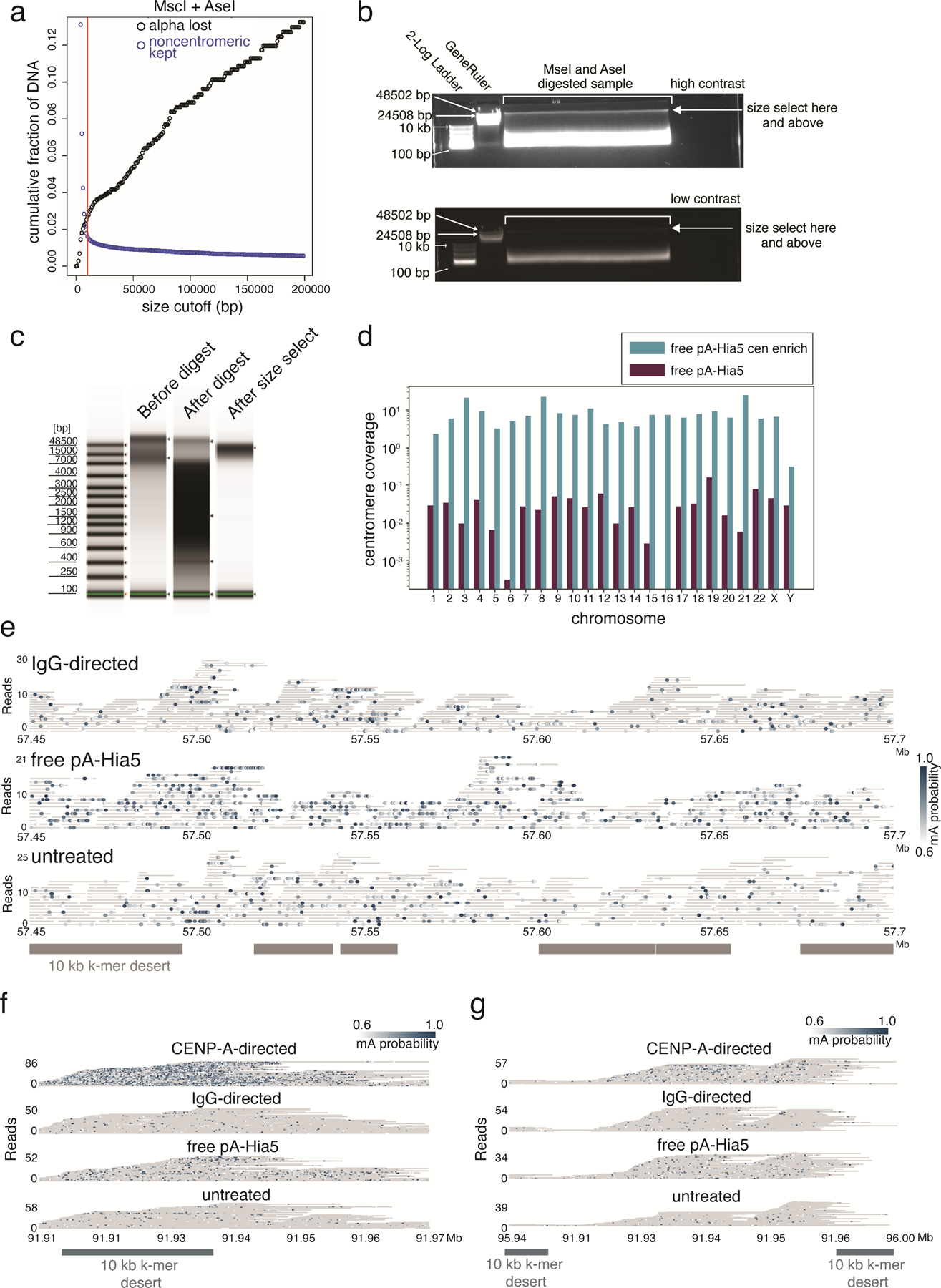
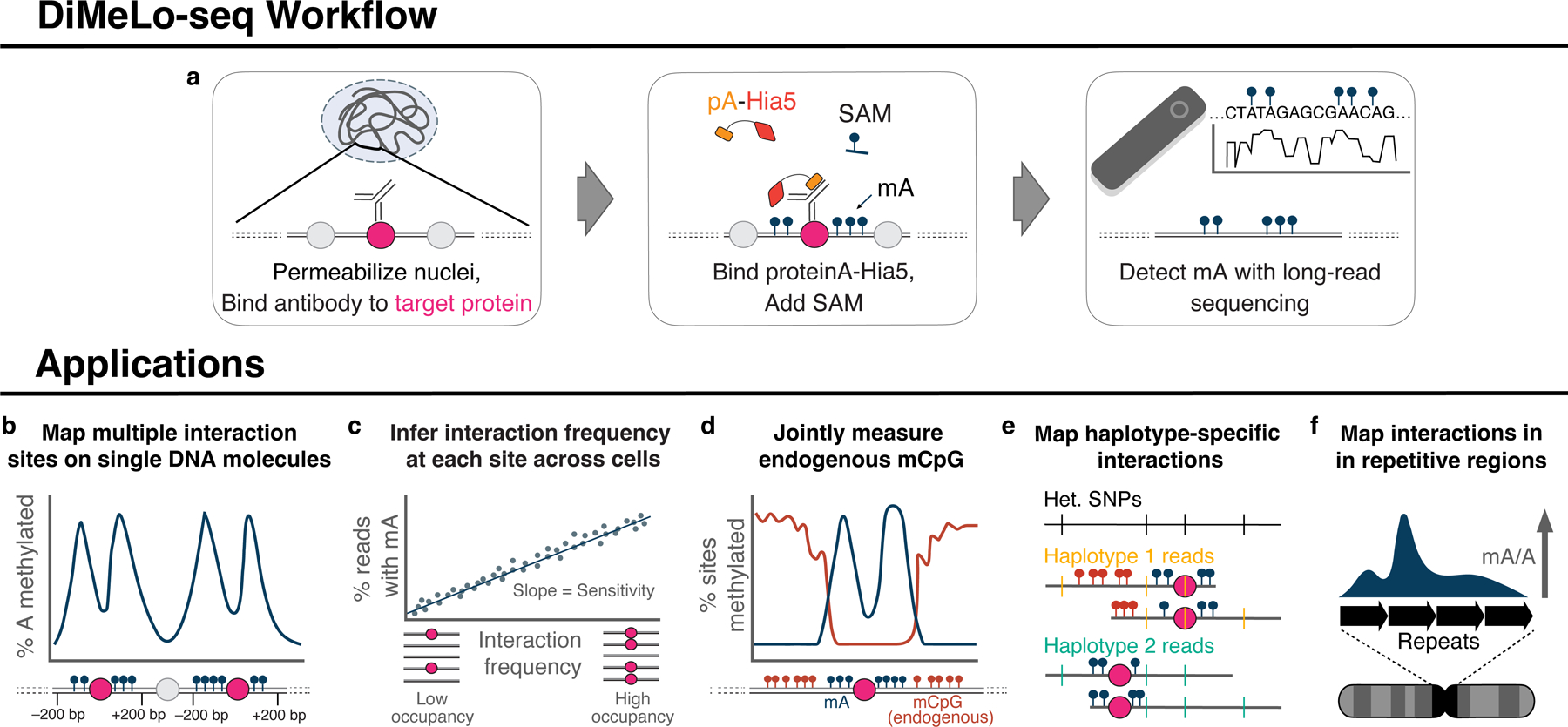
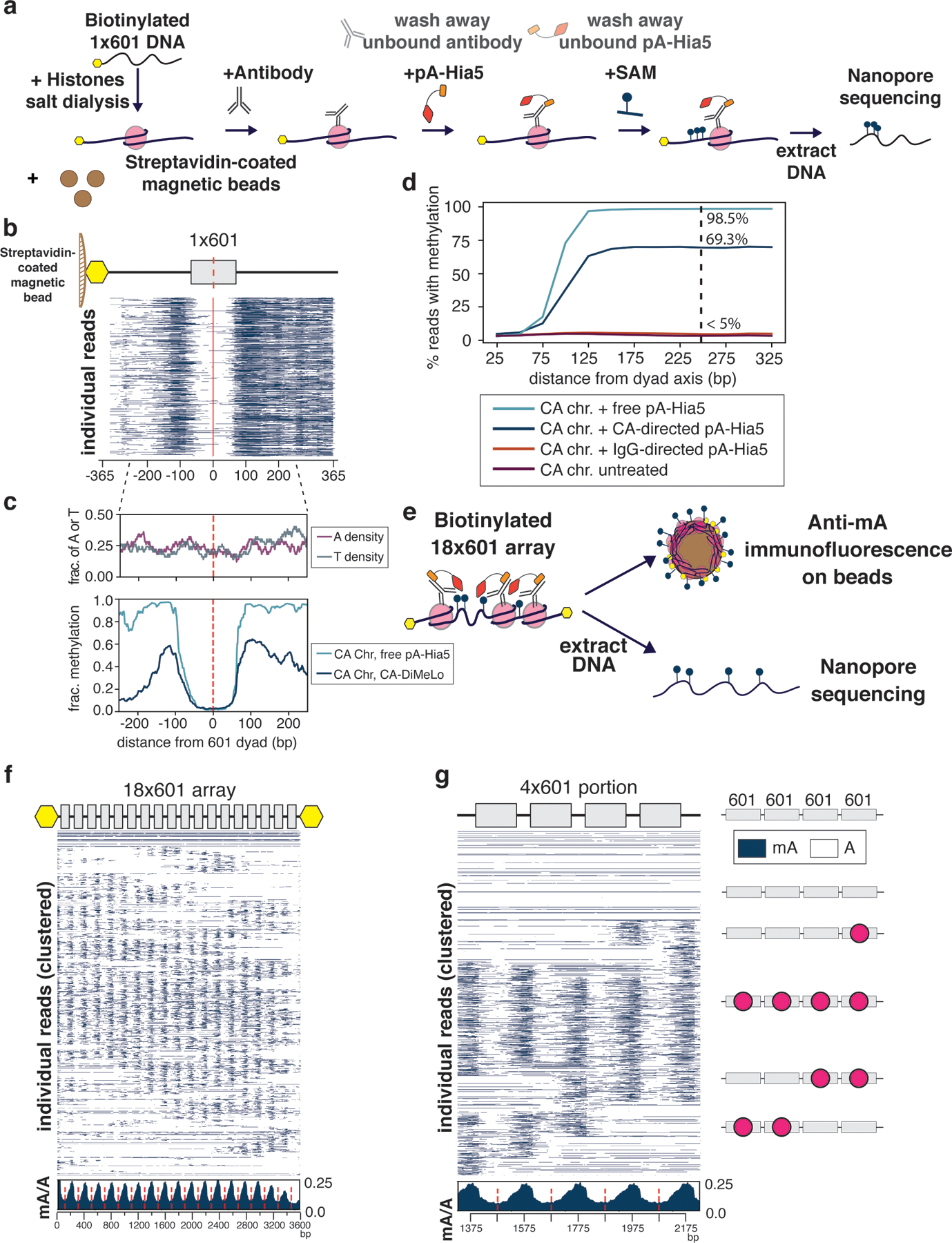
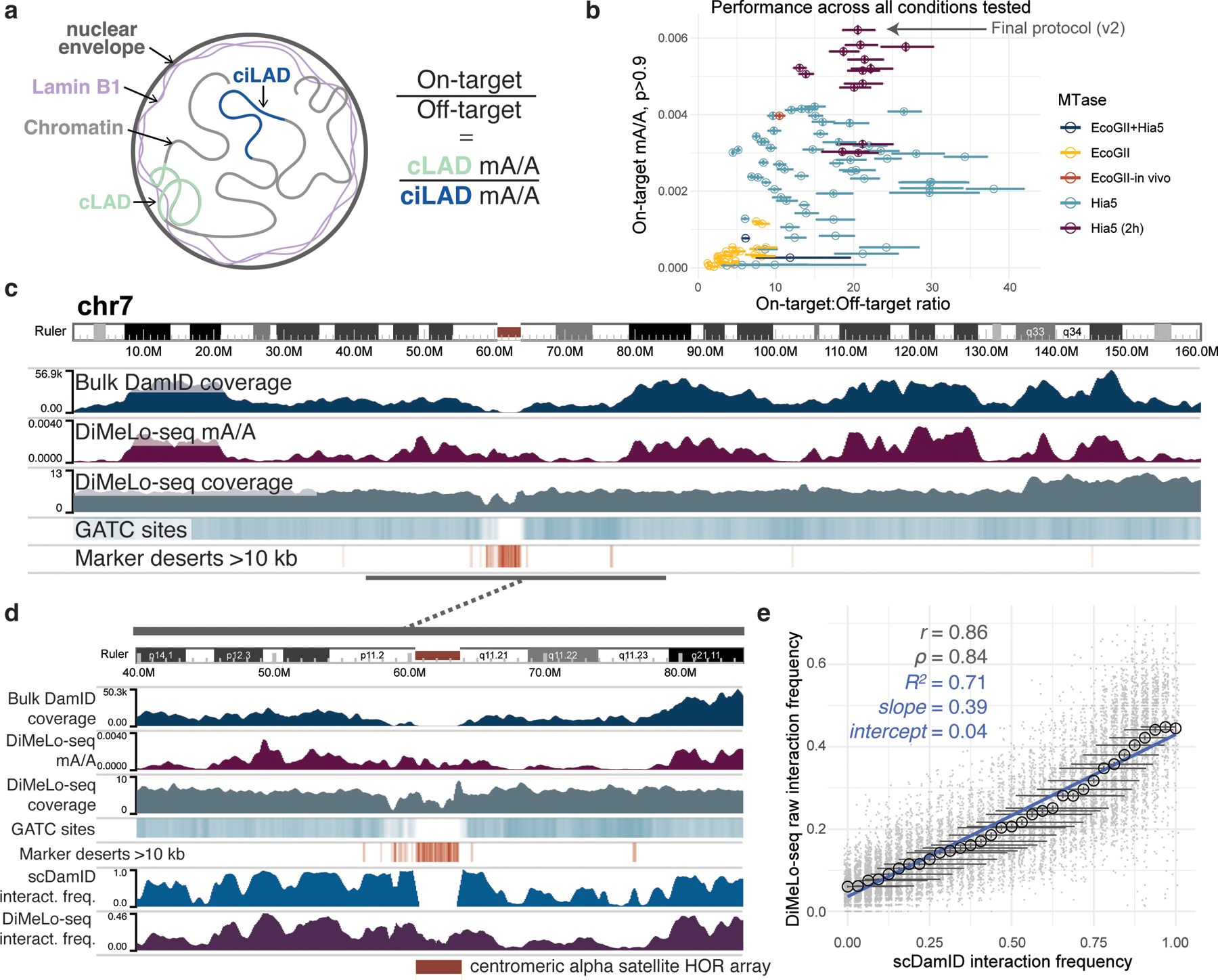
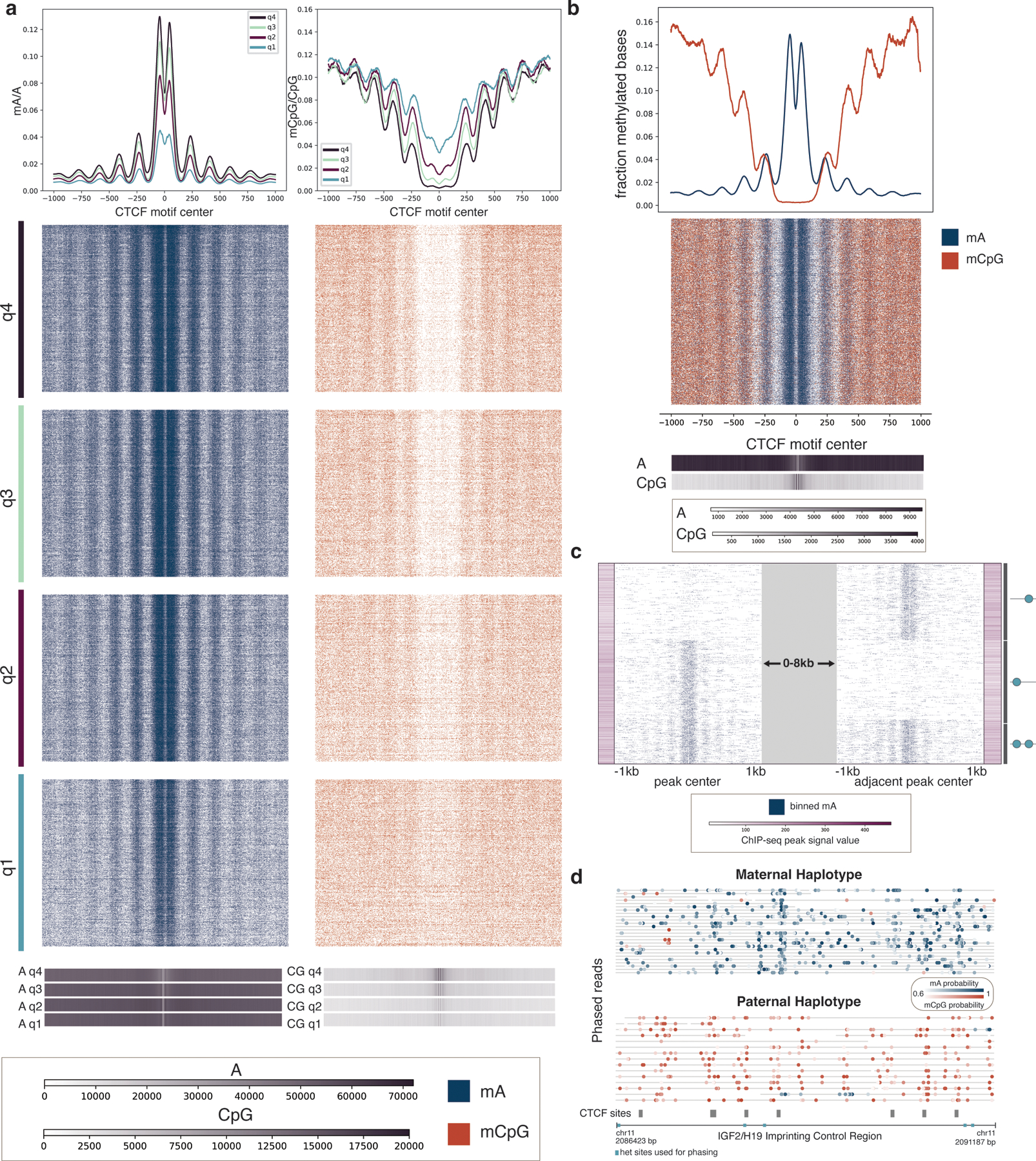
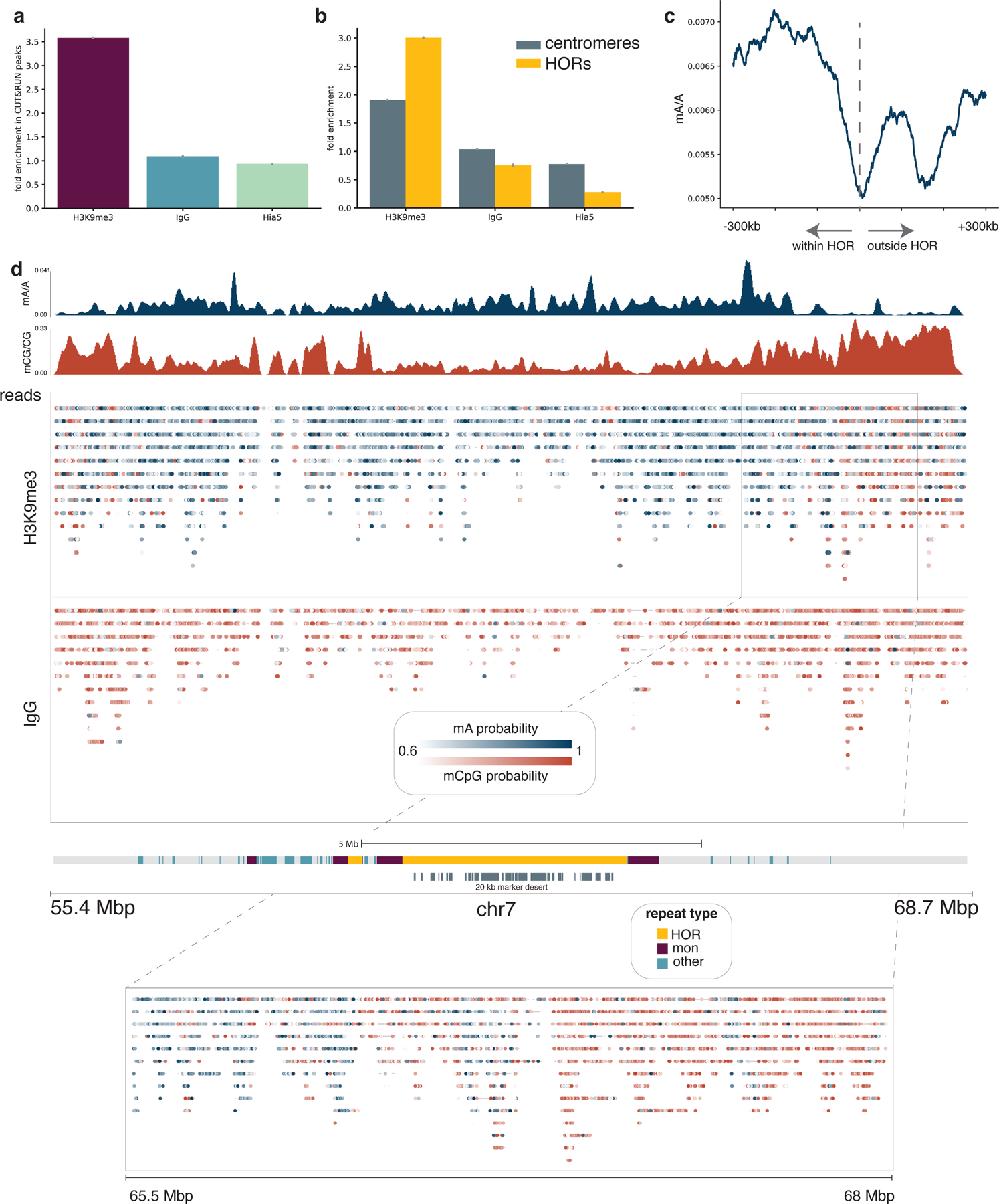
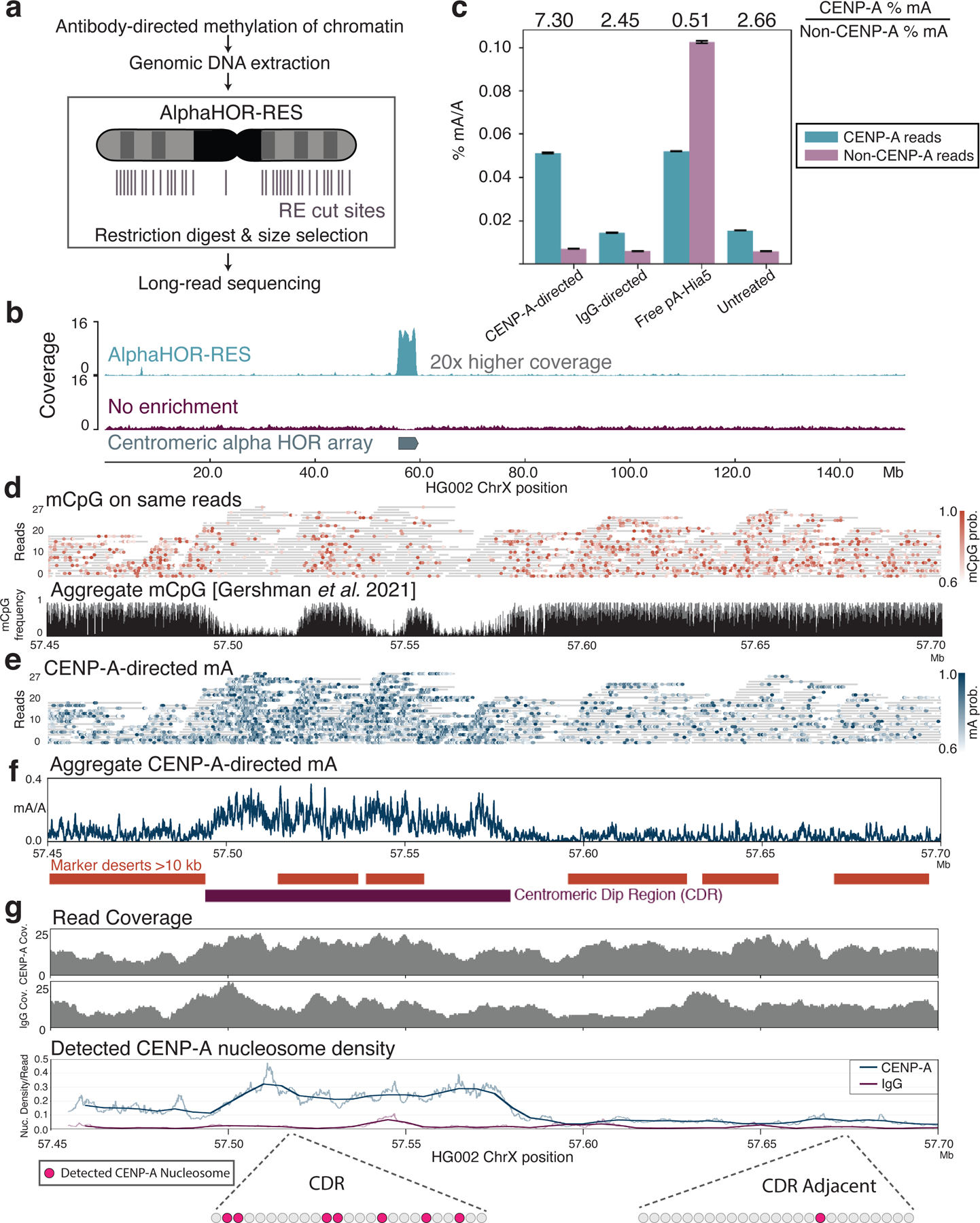
Studies of genome regulation routinely use high-throughput DNA sequencing approaches to determine where specific proteins interact with DNA, and they rely on DNA amplification and short-read sequencing, limiting their quantitative application in complex genomic regions. To address these limitations, we developed directed methylation with long-read sequencing (DiMeLo-seq), which uses antibody-tethered enzymes to methylate DNA near a target protein's binding sites in situ. These exogenous methylation marks are then detected simultaneously with endogenous CpG methylation on unamplified DNA using long-read, single-molecule sequencing technologies. We optimized and benchmarked DiMeLo-seq by mapping chromatin-binding proteins and histone modifications across the human genome. Furthermore, we identified where centromere protein A localizes within highly repetitive regions that were unmappable with short sequencing reads, and we estimated the density of centromere protein A molecules along single chromatin fibers. DiMeLo-seq is a versatile method that provides multimodal, genome-wide information for investigating protein-DNA interactions.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing Interests Statement
NA, AM, OKS, KS, AFS, and AS are co-inventors on a patent application related to this work. The remaining authors declare no competing interests.
Figures
References
-
- van Steensel B & Henikoff S Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat. Biotechnol 18, 424–428 (2000). - PubMed
-
- Robertson G et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 4, 651–657 (2007). - PubMed
-
- Johnson DS, Mortazavi A, Myers RM & Wold B Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502 (2007). - PubMed
-
- Barski A et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
