

Rearrangement processes and structural variations show evidence of selection in oesophageal adenocarcinomas
- PMID: 35396535
- PMCID: PMC8993906
- DOI: 10.1038/s42003-022-03238-7
Rearrangement processes and structural variations show evidence of selection in oesophageal adenocarcinomas
Abstract
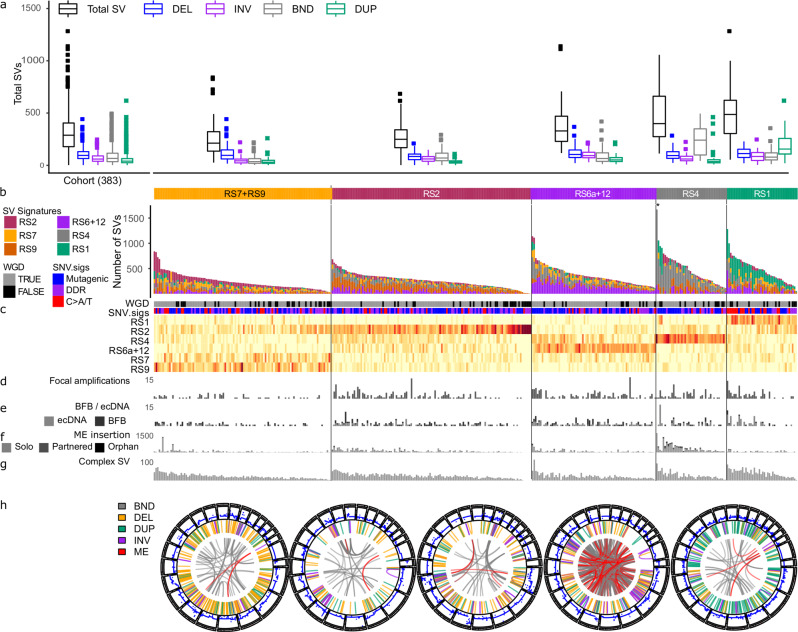
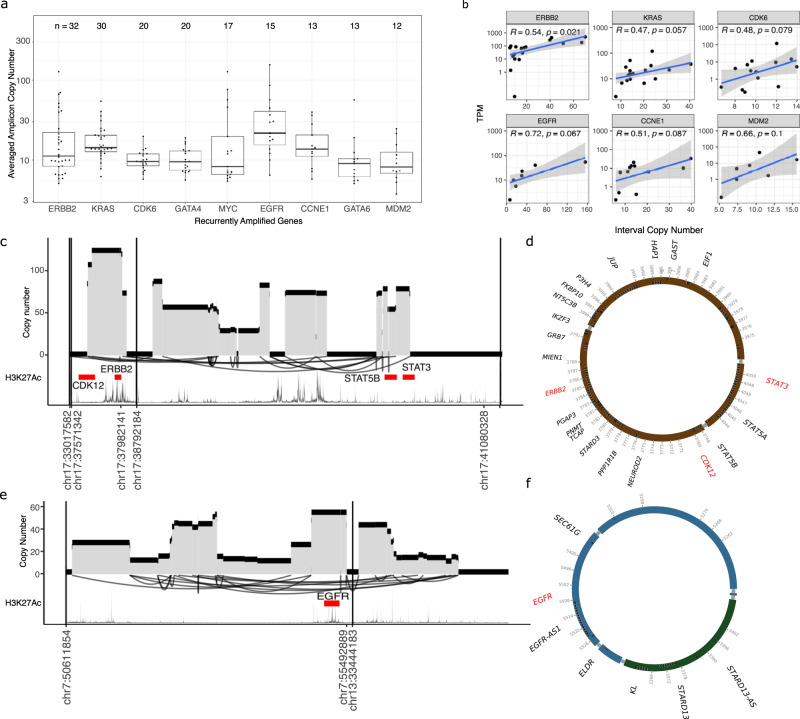
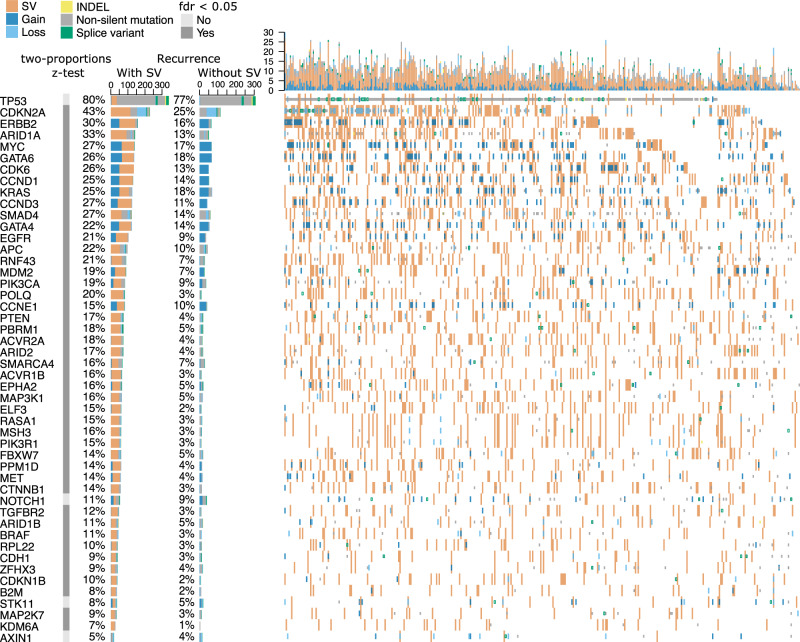
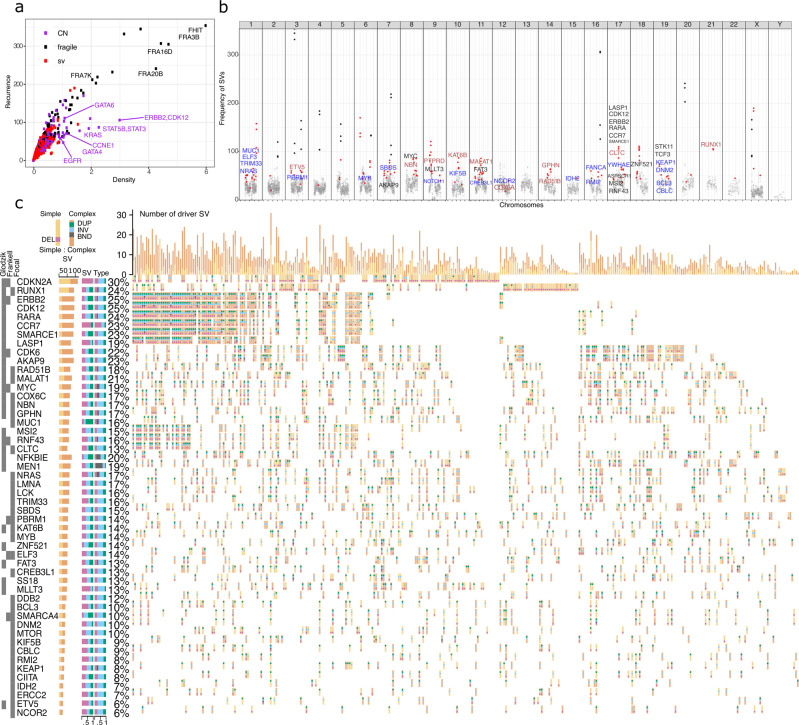
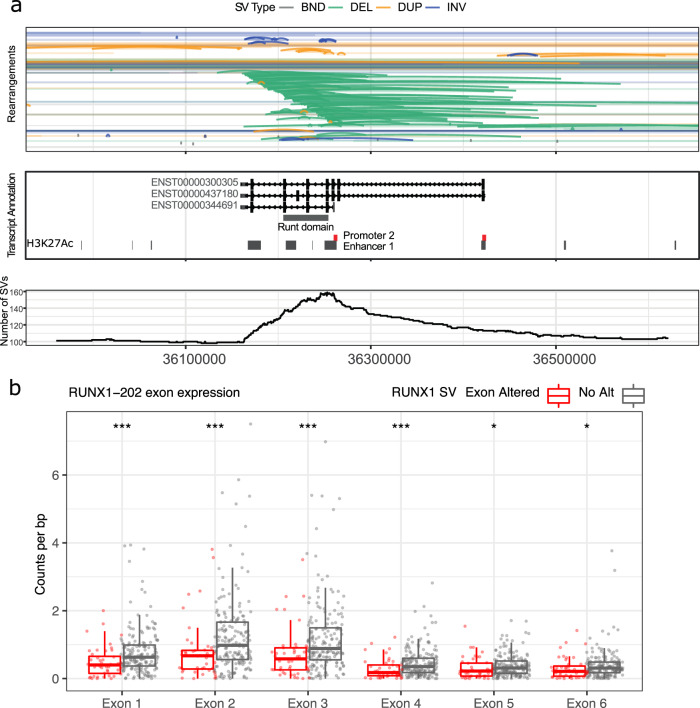
Oesophageal adenocarcinoma (OAC) provides an ideal case study to characterize large-scale rearrangements. Using whole genome short-read sequencing of 383 cases, for which 214 had matched whole transcriptomes, we observed structural variations (SV) with a predominance of deletions, tandem duplications and inter-chromosome junctions that could be identified as LINE-1 mobile element (ME) insertions. Complex clusters of rearrangements resembling breakage-fusion-bridge cycles or extrachromosomal circular DNA accounted for 22% of complex SVs affecting known oncogenes. Counting SV events affecting known driver genes substantially increased the recurrence rates of these drivers. After excluding fragile sites, we identified 51 candidate new drivers in genomic regions disrupted by SVs, including ETV5, KAT6B and CLTC. RUNX1 was the most recurrently altered gene (24%), with many deletions inactivating the RUNT domain but preserved the reading frame, suggesting an altered protein product. These findings underscore the importance of identification of SV events in OAC with implications for targeted therapies.
© 2022. The Author(s).
Conflict of interest statement
R.C.F. has devised an early detection technology called Cytosponge, this device technology and the associated TFF3 biomarker are licensed to Covidien GI solutions (now owned by Medtronic) by the Medical Research Council. R.C.F. and M.O. are named inventors on patents pertaining to the Cytosponge and associated technology. R.C.F. is a shareholder of Cyted Ltd., a company working on early detection technology. R.C.F. has received consulting and/or speaker fees from Medtronic, Roche and Bristol Myers Squibb. The other authors declare no competing interests.
Figures
References
-
- Sabarinathan, R. et al. The whole-genome panorama of cancer drivers. Preprint at bioRxiv10.1101/190330 (2017).
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
